CHIMICA sperimentale
Esperienze in home-lab: considerazioni di chimica sperimentale e altro
Messaggi di Aprile 2011
|
Post n°102 pubblicato il 28 Aprile 2011 da paoloalbert
Leggendo tra le righe con spirito critico i risvolti esclusivamente tecnici del lavoro descritto nel post precedente, rilevo un particolare poco chiaro nelle righe dell'Ambrosioni; forse si sarà trattato di un'omissione o di una svista dell'Autore, oppure di qualcosa che mi sfugge. |
|
Post n°101 pubblicato il 26 Aprile 2011 da paoloalbert
Dopo le quattro puntate precedenti, non val la pena di rilassarsi un pochino leggendo un paio di paginette dell'Ambrosioni, come quando si va in bagno e si cerca qualche lettura di poco impegno?
-L'azzurro di Berlino conosciuto in commercio altresì coi nomi di bleu, ossia azzurro Prusso, e dai Chimici Idrocianato di tritossido di ferro, trae il suo nome dalla città, ove per la prima volta è stato inventato, e fabbricato. |

|
Post n°100 pubblicato il 23 Aprile 2011 da paoloalbert
I don't know how to love him... -Non so come amarlo- cantava la struggente Ivonne Elliman nel 1970 in uno dei più significativi momenti dell'opera pop Jesus Christ Superstar.
|
|
Post n°99 pubblicato il 19 Aprile 2011 da paoloalbert
Dopo essere stato preventivamente testato al banco mentre cresceva (ved. foto nel post precedente), ho slegato dal lettino il circuito "piccolo Frankestein" liberandolo dalla miriade di fili che lo alimentavano e l'ho piazzato in una scatoletta che avevo, in modo che fosse indipendente e potesse cominciare a muoversi autonomamente facendo il lavoro che doveva fare.
Le foto, fatte in qualche modo di sera, rendono un'idea del marchingegno finito durante una misura sotto descritta.
Prove di assorbimento |


|
Post n°98 pubblicato il 17 Aprile 2011 da paoloalbert
Dopo il lavoro di precisione abbastanza noioso ma indispensabile per la celletta, mi sono preso un po' più di soddisfazione con la progettazione e realizzazione del circuito. Sottolineo che occorre amplificare non il segnale, ma le "variazioni" del segnale stesso a seconda della concentrazione, e queste variazioni possono essere debolissime, anche se il segnale in assoluto è forte.
Nella foto si vede l'accrocco mentre è ancora in fase di progettazione, i componenti disassemblati belli distesi e pieni di fili come Frankestein, che dà i primi segni di vita leggendo l'assorbimento di una soluzione di sali di rame.
Le prime prove all'oscilloscopio mostrano che l'apparecchio come principio funziona e sono quelle che mi danno la forza di continuare nella sperimentazione! |



|
Post n°97 pubblicato il 14 Aprile 2011 da paoloalbert
Come dicevo nell'introduzione, tempo fa mi è venuta voglia di provare a fare un piccolo "spettroscopio/colorimetro" sperimentale, naturalmente senza alcun intendimento di costruire uno strumento di misura, ma con lo scopo di fare un "giocattolo", diciamo così, didattico che verificasse l'assorbanza relativa di uno ione (o di una molecola colorata) e anche per avere la scusa di sposare bene assieme i miei due hobbies principali (chimica ed elettronica).
La cuvetta è quel piccolo contenitore parallelepipedo di cui è noto il cammino ottico ed il materiale è di buona trasparenza; ci sono cuvette in vetro, a facce perfettamente parallele e calibrate (molto costose), e cuvette economiche in plastica: indovinare quali ho usato io... Le due protuberanze laterali in rame che si vedono in foto contengono uno l'elemento illuminatore e l'altro il ricevitore.
|

|
Post n°96 pubblicato il 10 Aprile 2011 da paoloalbert
Cercando in rete l'argomento "spettroscopia UV-visibile" si trova tutto quello che io ometterò di dire in questa sede, la quale, come ormai ben si sa, è dedicata quasi categoricamente alla realizzazione pratica di quello che la teoria propone. Solo due parole minime introduttive al mio esperimento (anche hardware!) che seguirà: la spettroscopia è una disciplina che riguarda quelle tecniche con le quali è possibile ottenere informazioni sulle proprietà strutturali dei corpi studiando l’interazione della materia con l’energia elettromagnetica, cioè la luce, visibile o meno.
Dopo questa doverosa premessa ULTRA-semplificata, ho provato a costruire un aggeggio che facesse vagamente quanto sopra: |
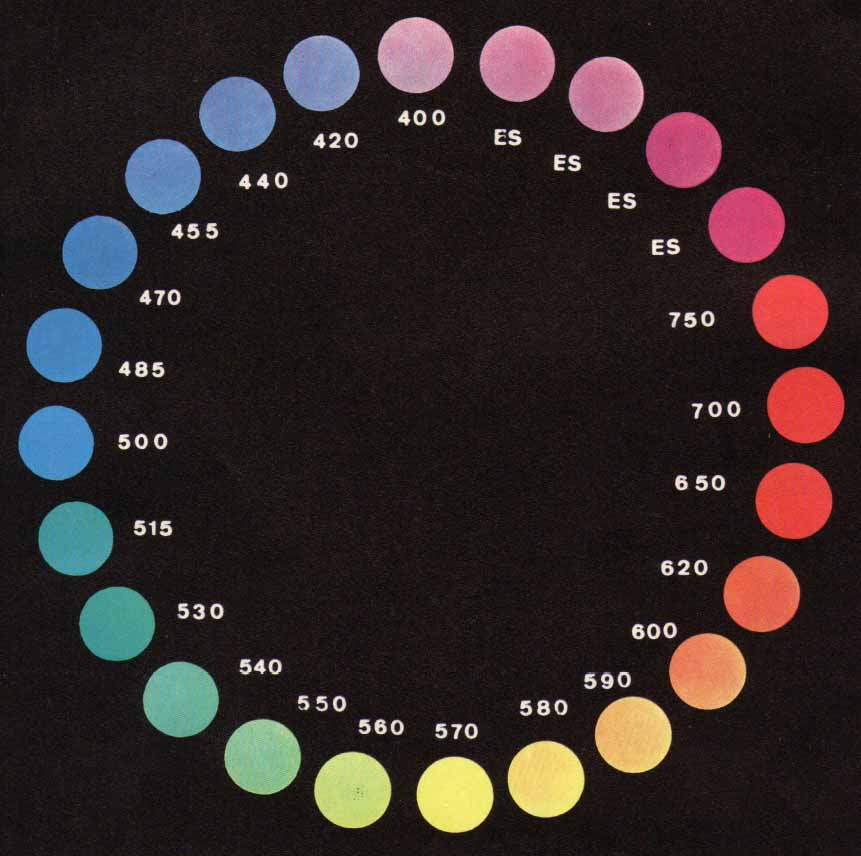 Se un raggio di luce policromatica illumina un oggetto che ne può assorbire una parte, la radiazione che giunge all’occhio contiene solo le lunghezze d’onda che non sono state assorbite, cioè le radiazioni complementari.
Se un raggio di luce policromatica illumina un oggetto che ne può assorbire una parte, la radiazione che giunge all’occhio contiene solo le lunghezze d’onda che non sono state assorbite, cioè le radiazioni complementari.|
Post n°95 pubblicato il 05 Aprile 2011 da paoloalbert
Nel blog ci vuole ogni tanto uno stacchetto, magari d'arte (una musica, un'immagine...).
|


|
Post n°94 pubblicato il 03 Aprile 2011 da paoloalbert
Ironia della sorte, una volta abitavo vicino ad una località che si chiamava proprio "Silani"... una specie di microscopica Silicon Valley ante litteram: ma quelle quattro case non avevano a che fare con i composti del silicio! |
Inviato da: paoloalbert
il 02/05/2021 alle 21:53
Inviato da: Bepi1249
il 15/04/2021 alle 14:11
Inviato da: Tanner85
il 06/02/2021 alle 00:01
Inviato da: poetryclub
il 04/10/2020 alle 23:00
Inviato da: poetryclub
il 04/10/2020 alle 20:58