CHIMICA sperimentale
Esperienze in home-lab: considerazioni di chimica sperimentale e altro
|
Post n°193 pubblicato il 18 Agosto 2012 da paoloalbert
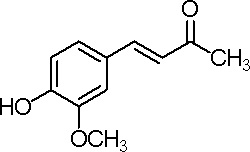
Il nome altisonante di questa sintesi non rende giustizia alla sua discreta semplicità; è una sintesi carina e di soddisfazione nei risultati, anche se alla fine c'è "un'inezia" che personalmente avrei preferito non ci fosse: il doppio legame nella molecola!
Si tratta di una condensazione aldolica (ved. altrove), simile a quella presentata tempo addietro tra l'acetone e la benzaldeide per la sintesi del dibenzalacetone.
Dopo qualche minuto, se non si agita, si nota la formazione sul fondo di liquido rosso che si separa ma che si ridiscioglie semplicemente agitando.
Il solido filtrato si presenta microcristallino e di color verdino pallido.
Coprire il becher con vetrino d'orologio e lasciar raffreddare lentamente ed in riposo per qualche ora, fino a cristallizzazione completa.
|






|
Post n°192 pubblicato il 06 Agosto 2012 da paoloalbert
Mi piacciono le sostanze particolarmente "reattive": sono attrici dinamiche e vivaci, sulle quali si può contare quando c'è da imbastire una bella commediola che abbia come trama una sintesina di chimica organica.
Nel mio caso la "cappa" è costituita da una finestrella che si apre direttamente sul tetto dell'edificio dove ho il lab e che quasi sempre produce una utilissima corrente d'aria aspirante verso l'alto, come una perfetta cappa naturale.
Quando tutto l'acido è stato introdotto nel pallone, scaldare a riflusso per mezzo di un bagno d'acqua a 60-65° finchè non si ha più emissione di fumi bianchi (circa un'ora). Sostituire l'allhin con un condensatore Liebig e distillare raccogliendo fino a circa 65°
Il residuo, c
Questo cloruro acilico si presenta come un liquido incoloro mobilissimo fumante all'aria (si idrolizza istantaneamente ad acido acetico ed acido cloridrico), di odore estremamente acre ed irritante, corrosivo, da maneggiare con molta cautela ma utile in tante reazioni di acetilazione. |
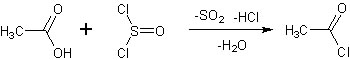
 In un pallone a due colli da 250 ml porre 80 ml di
In un pallone a due colli da 250 ml porre 80 ml di 

 ontenente chissà quali sottoprodotti clorurati, è estremamente irritante e lacrimogeno.
ontenente chissà quali sottoprodotti clorurati, è estremamente irritante e lacrimogeno. 
|
Post n°191 pubblicato il 01 Agosto 2012 da paoloalbert
Gli alogenuri acilici sono reagenti fondamentali per la chimica organica per una nutrita serie di sostituzioni nucleofile, che avvengono ognuna nelle condizioni adatte. La prossima volta vedremo come. |
|
Post n°190 pubblicato il 25 Luglio 2012 da paoloalbert
|

|
Post n°189 pubblicato il 18 Luglio 2012 da paoloalbert
Premetto subito, avendola ripetuta più volte cocciutamente, che la preparazione di questo reattivo per il sodio è comunque di bassissima resa, con esiti nel mio caso mai andati completamente a buon fine.
Precipitato fioccoso (acido antimonico?) che si forma in varie fasi della sperimentazione con il piroantimoniato. Questo NON è il piroantimoniato sodico, del tutto diverso. Seccando il prodotto si ottiene una polvere del tutto insolubile ed inutile.
Prova in bianco: soluzione molto diluita di piroantimoniato; si nota comunque un principio di idrolisi, nonostante la forte alcalinità residua.
Aggiunta di sol. di NaCl: prec. microcristallino: finalmente! |



|
Post n°188 pubblicato il 14 Luglio 2012 da paoloalbert
Se dovessi dire qual'è la preparazione chimica che più mi ha fatto tribolare direi che quella del piroantimoniato la metterei prima in classifica. La prossima volta tenterò, sperando nell'aiuto di queste buone anime... |
|
Post n°187 pubblicato il 03 Luglio 2012 da paoloalbert
Fine giugno - primi luglio 2012: caldo tropicale sull'Italia... Aspettando il prossimo post, nell'intervallo ci sta questa vecchia immagine presa quando sperimentavo con le celle di Peltier: un bel becher fresco sudante che vorrebbe trasformarsi in acqua ghiacciata...
Evviva il caldo, evviva il freddo!
|

|
Post n°186 pubblicato il 27 Giugno 2012 da paoloalbert
Questo dovrebbe essere l'ultimo lavoro riguardo la saga della silice blù, e l'inizio del periodo annuale di rarefazione dei post su questo blog. |
|
Post n°185 pubblicato il 21 Giugno 2012 da paoloalbert
Sono convinto che la realtà superi sempre la fantasia. |
|
Post n°184 pubblicato il 16 Giugno 2012 da paoloalbert
Il mistero del colorante blù del gel di silice continua... -"Come primaaa, più di primaaaa...", cantava una volta Tony Dallara!
Una volta arrivati a secco... ecco la prima sorpresa: un bel residuo inaspettato, color beige.
|



|
Post n°183 pubblicato il 13 Giugno 2012 da paoloalbert
Quando si ha a che fare con l'elettrochimica prima o poi non si scappa: si ha il problema del "vaso poroso"! E' un elemento indispensabile quando le soluzioni in gioco sono due (come spesso accade), le quali non devono mescolarsi ma deve essere consentito uno scambio ionico tra di esse. L'esempio classico sono le pile Daniell, Bunsen o Leclanchè, dove i due metalli sono nella rispettiva semicella separati appunto da un introvabile "vaso poroso". Anch'io non sapevo mai come risolvere questo problema... fino a ieri, quando sono riuscito inaspettatamente a farmi fare da un artigiano della ceramica il cilindretto che si vede in foto. Finalmente potrò tentare un esperimento di elettrochimica al quale pensavo da tempo e qualcos'altro che mi verrà in mente.
|

|
Post n°182 pubblicato il 08 Giugno 2012 da paoloalbert
I lettori più affezionati di questo blog ricorderanno che nei post dal 96 al 99 si parlò di un "C O L O R I M E T R O" che avevo costruito quasi per gioco, in una delle mie alternanze ricorrenti tra hobby chimico ed hobby elettronico.
Le due "protuberanze" in tubetto di rame che si vedono nell'immagine contengono rispettivamente un diodo LED di illuminazione (a destra) ed un fototransistor di lettura (a sinistra).
Naturalmente tutto questo è stato fatto per gioco (vogliamo essere più generosi? Diciamo allora per ricerca personale...) e non vi è nessuna velleità di misurazioni quantitative nè di impiego pratico di questo "colorimetro alla mia maniera". Ma rimane molto forte e impagabile, questa sì, la soddisfazione di aver fatto (e imparato!) qualcosa abbinando la teoria con la pratica, la chimica con l'elettronica... mixando semplicemente un paio di hobbies. Cosa si vuole di più? |

|
Post n°181 pubblicato il 03 Giugno 2012 da paoloalbert
Una immagine vale come mille parole... si dice, ed è vero.
|

|
Post n°180 pubblicato il 30 Maggio 2012 da paoloalbert
Segue dalla puntata precedente. I gr
Allontano a questo punto tutto il silicio per ebollizione prolungata a bagno maria e ottengo un concentrato con ancora molto HF in eccesso, nella soluzione più altobollente di 100°. Voglio tirare tutto a secco, come fare? Non potendo adoperare per ovvie ragioni oggetti in vetro (nè tantomeno crogiolini in platino!), preparo per l'occasione un piccolo evaporatore in teflon fresandone una cavità in un pezzetto di questo materiale; il teflon può essere riscaldato anche ad alta temperatura e resiste perfettamente all'acido fluoridrico.
Una volta arrivato a secchezza... sorpresa, ecco il risultato! Un bel po' di mg di residuo perfettamente gestibili, ma non di un bel colore azzurro come mi sarei maggiormente aspettato, bensì di un sospetto color ruggine.
Non sarà mica ferro anche stavolta! (ormai il ferro mi sta stufando quando salta fuori mentre mi aspetto altri elementi!).
Test con tiocianato - Test con ferrocianuro
Test con K etilxantato - Test con tracce di rame aggiunte
Conclusioni
Chissà quale prodotto cinese a base di ferro, forse ftalocianine o qualcosa di simile? |







|
Post n°179 pubblicato il 26 Maggio 2012 da paoloalbert
No, non mi è dato di volta il cervello (almeno non del tutto...), ma il discorso che segue è veramente dedicato a un gatto, quello della gentile Teresa (alla quale mi permetto invece di dedicare il quadro di Renoir...).
|

|
Post n°178 pubblicato il 18 Maggio 2012 da paoloalbert
Dopo un paio di post "storici", devo mantenere la promessa di mettere mano alla vetreria!
I componenti
Lo scopo di questo lavoro era verificare i parametri tensione/corrente della pila e quindi è indispensabile almeno un tester affidabile; ho usato il multimetro Schlumberger che si vede in seguito nelle immagini.
|




|
Post n°177 pubblicato il 14 Maggio 2012 da paoloalbert
Anche oggi un po' di storia... la prossima volta metto mano alla vetreria, promesso!
Monsieur Eugene Grenet per la verità ha fatto un po' troppo il furbo, perchè è dal 1856 che si prende tutto il merito per aver dato il nome a questa pila, che in realtà era stata inventata già quattordici anni prima da Johann Christian Poggendorff; lui l'ha resa solo un po' più pratica, prendendosi pure un brevetto tre anni più tardi. Questa pila, come quella di Volta, è un generatore ad un solo conduttore di seconda classe, cioè di una pila in cui la soluzione elettrolitica è una sola (conduttori di prima classe sono gli elettrodi metallici, di seconda la soluzione ionica). Grenet sostituì questo composto, scomodo da ottenere puro, con una soluzione solforica di bicromato di potassio K2Cr2O7, che è poi la stessa cosa (l'anidride cromica si ottiene per azione dell'H2SO4 sul bicromato; è la separazione successiva della CrO3 pura che è difficile). Ecco spiegato come gli elettrodi di carbone non partecipino alla reazione (non si consumano), mentre la povera lamina di zinco deve ossidarsi prendendosi sulle spalle metà del lavoro e sciogliendosi lentamente nella soluzione come solfato di zinco ZnSO4. La disposizione degli elettrodi, molto vicini e di grande superficie, unita al tipo di elettrolita, conferisce alla pila una resistenza interna particolarmente bassa e la rende quindi idonea ad erogare correnti molto intense, anche se per poco tempo. Per quanto affidabili e capaci di produrre un'intensa corrente, le pile al bicromato sono andate in disuso verso la fine del XIX secolo perché tendevano a "scaricarsi" troppo velocemente, anche se lo zinco veniva inserito soltanto quando si voleva alimentare il circuito utilizzatore.
|


|
Post n°176 pubblicato il 08 Maggio 2012 da paoloalbert
L'amico Marco mi ha recentemente invitato ad assistere ad una bella (quanto inconsueta!) conferenza sulla "Breve storia delle lampade da minatore"... ghiotta occasione alla quale ho dovuto con rammarico rinunciare.
---°°°OOO°°°--- |

|
Post n°175 pubblicato il 04 Maggio 2012 da paoloalbert
Oggi niente divagazioni storico-personali: lo chef ci schiaffa in tavola un piatto di sola chimicaccia per palati allenati. Ogni tanto una portata pesante rende più gradevoli le successive...
Materiale occorrente:
Il 1-naftalensulfonato sodico si presenta sotto forma di polvere microcritallina leggera che tende a impaccarsi di colore appena appena beige; non deve odorare per niente di naftalina. |
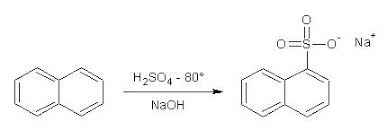
 Il naftalene tende a sublimare facilmente e nelle zone fredde del pallone si formerà uno straterello di sostanza sublimata, che non parteciperà alla reazione.
Il naftalene tende a sublimare facilmente e nelle zone fredde del pallone si formerà uno straterello di sostanza sublimata, che non parteciperà alla reazione.  In ogni caso filtrare, ottenendo una soluzione solo leggermente opalescente.
In ogni caso filtrare, ottenendo una soluzione solo leggermente opalescente.  Scaldare fino a circa 80° eventualmente continuando ad aggiungere NaCl alla soluzione che a caldo rimane leggermente rosata ma quasi limpida.
Scaldare fino a circa 80° eventualmente continuando ad aggiungere NaCl alla soluzione che a caldo rimane leggermente rosata ma quasi limpida. 
|
Post n°174 pubblicato il 27 Aprile 2012 da paoloalbert
A Montevecchio ero riuscito fortunosamente a reperire, come si ricorderà, due "sassi" interessanti, gli unici fortemente mineralizzati che ero riuscito a trovare razzolando in una discarica difficilmente accessibile.
Devo dire che l'analisi ha completamente smentito le mie supposizioni: avevo ritenuto trattarsi di blenda... e invece di tutt'altro si tratta! Ma andiamo a procedere. Ho trattato 5 g di minerale, scegliendo la parte con meno ganga, con acido cloridrico concentrato a caldo, fino a dissoluzione quasi completa.
Precipitazione del cromato di piombo Il piombo infatti, se in piccola quantità, non rimane tutto come residuo insolubile del primo gruppo analitico ma passa in soluzione anche sotto forma del poco solubile PbCl2 e viene rivelato con sicurezza dal cromato.
Ferrocianuro ferrico (Blù di Prussia) Non metto le formule e le reazioni perchè sono banali e scontate; ecco le due foto finali, del tiocianato ferrico e del blù di Prussia, che dichiarano a gran voce che il minerale trovato NON era blenda ma un volgare (per modo di dire) minerale di ferro contaminato, diciamo così, da una discreta quantità di galena con associata pochissima blenda. |




Inviato da: paoloalbert
il 02/05/2021 alle 21:53
Inviato da: Bepi1249
il 15/04/2021 alle 14:11
Inviato da: Tanner85
il 06/02/2021 alle 00:01
Inviato da: poetryclub
il 04/10/2020 alle 23:00
Inviato da: poetryclub
il 04/10/2020 alle 20:58