CHIMICA sperimentale
Esperienze in home-lab: considerazioni di chimica sperimentale e altro
|
Post n°333 pubblicato il 06 Novembre 2015 da paoloalbert
A proposito di litio, scommetto che tanti giovani studenti con qualche pallino per la chimica hanno visto il colore alla fiamma di questo metallo solo nelle immagini di Google. a)- si prendeva una bacchetta di vetro e fondendone parzialmente una estremità sul bunsen le si inseriva per qualche millimetro un filetto di platino. b)- siccome il filetto era stato toccato dalle manacce dello studente (o dell'Assistente) lasciandone tracce di sodio sulla superficie, lo si immergeva in acido cloridrico concentrato e lo si passava alla fiamma più e più volte, finchè la fiamma medesima non avesse perso quella insistentissima colorazione gialla. c)- si metteva il filo nella fiamma ossidante e quasi incolore del bunsen e se ne osservava il colore, memorizzandolo per l'eternità. d)- dopo ogni osservazione si puliva di nuovo accuratamente il filo con i soliti bagnetti acidi (perchè si usasse il platino mi sembra superfluo dire: perchè è un metallo nobilissimo, inalterabile e resiste quasi a tutto. Perchè si usasse proprio l'acido cloridrico per queste prove è per il fatto che i cloruri dei metalli sono più "volatili" degli altri sali, senza avere zavorre anioniche attaccate al metallo). e)- quando si arrivava al litio... -che bello!- tutti esclamavano! -E' il rosso cardinale del litio- diceva il paleozoico insegnante.
La zona ossidante della fiamma è quasi incolore
Il rosso cardinale del litio
Ma perchè certi metalli colorano la fiamma? Si tratta di elettroni che saltando da un'orbitale all'altro eccitati dal calore emettono fotoni su una certa lunghezza d'onda... ma vedo che sto deviando e mi fermo immediatamente: se cominciamo a parlare di orbitali ed elettroni non siamo più in Chimica sperimentale. -I fondi non ci sono, le sostanze sono pericolose, non c'è tempo per queste cose, il metano è pericoloso, non abbiamo gli assistenti, le scorte di reagenti sono finite, il Ministero non ci manda più i soldi, meglio fare materie umanistiche... eccetera, eccetera, eccetera all'infinito- |


|
Post n°332 pubblicato il 21 Ottobre 2015 da paoloalbert
Ai margini del bosco, a due passi da casa, c'è questo vecchissimo melo semiselvatico, che fa delle piccole mele verdi e rosse di quelle qualità antiche ormai quasi scomparse.
|




|
Post n°331 pubblicato il 13 Ottobre 2015 da paoloalbert
Fame commerciale intendo... quante apparecchiature ci sono oggi nel mondo alimentate con una batteria al litio? Possiamo dire infinite?
- sodio 93 dove si vede che la quantità di litio (un elemento non certo comune) è eccezionalmente elevata; salta anche all'occhio l'assenza quasi totale del calcio (appena 0,03 g/l).
La separazione dei vari componenti è laboriosa ed avviene per lo più per cristallizzazione frazionata; la separazione del boro si effettua con solventi organici. Altri enormi giacimenti di litio in acque salate si trovano in Argentina (Jujuy) e in Nevada (Silver Peak), tutti siti gestiti da potenti multinazionali americane, che fanno il prezzo mondiale dell'elemento numero TRE nella lotta per aggiudicarsi un pezzo del monopolio di un elemento che sta diventando strategico. |



|
Post n°330 pubblicato il 29 Settembre 2015 da paoloalbert
Conosco una persona che odia le code, e le fa praticamente solo negli Uffici dello Stato, dove in nessun modo le può evitare. - Alle ore 09:20, quaranta minuti prima dell'apertura dei padiglioni, sono già dentro l'Expo! Non sono in ritardo, non temo nulla! - La mia entrata (porta Fiorenza) conduce proprio accanto al padiglione Zero, bene! - Ho sentito dire che il padiglione del Kazakistan è bello, assolutamente da non perdere; ottimo, dico, prima che ci sia troppa gente mi ci dirigo, anche se so che sarà lontano. - Per farla breve, dopo un bel po' arrivo al Kazakistan (durante la passeggiata ero entrato ed uscito in 30 secondi dal padiglione del Camerun, dove avevo trovato la copia esatta, o quasi, di quelle belle statuette in legno scuro esotico vendute d'estate su tutte le spiagge italiane dagli amici africani. Mi ero detto che magari la statuetta lunga e magra me l'avrei presa l'estate prossima a Cesenatico). - Sento da voci che girano che per il Giappone si parla di sette ore, per la Germania di cinque, altrettante per il padiglione Italia e altrettante per lo Zero della mattina, da quale ero partito. - Stranamente in dieci minuti entro anche nel padiglione francese, tutto pentole e ammenicoli appesi. - Durante la giornata faccio mille chilometri in su e in giù lungo il decumano, entrando di quà e di là dove la coda è inesistente o di pochi minuti. - E' notte, c'è il gioco di acqua e luci all'Albero della Vita, e io sono in prossimità... almeno questo spettacolo me lo guardo, non devo entrare da nessuna parte! - Vado ai padiglioni Mediterranei, dove non c'è (relativamente) nessuno. Una meraviglia. - Ormai è notte, tutti sloggiano. E' ora di far fare alle suole ormai consunte gli ultimi chilometri per tornare al parcheggio, levare le ancore e tornare a casa. Ecco la cronaca, molto riassunta, della visita all'Expo di quel tale che odiava le code (e che adesso odia ancora di più), che io fedelmente riporto come me l'ha raccontata.
|
|
Post n°329 pubblicato il 18 Settembre 2015 da paoloalbert
Ne ho visti di tramonti in montagna, di tutti i tipi direi. La fotografia è presa guardando ad ovest, naturalmente dopo il classico temporale, i residui del quale si trovano ora sopra di noi, e tutto è quasi nero.
Una immagine che non può nemmeno vagamente rendere giustizia alla realtà e quattro parole per un tramonto raro.
|

|
Post n°328 pubblicato il 28 Agosto 2015 da paoloalbert
Ciò che vado a descrivere è una via di mezzo tra una vera sintesi, con la sua bella procedura particolareggiata, e un "esperimento", giocato con modalità più semplificate.
E se ci fossero più nitrogruppi? |




|
Post n°327 pubblicato il 19 Agosto 2015 da paoloalbert
Visto che bel becco... Bunsen? |

|
Post n°326 pubblicato il 02 Agosto 2015 da paoloalbert
"I tempi sono cambiati... ma il libro rimane pregevole ed interessante..."
Il volume è diviso in cinque principali capitoli, scritti in forma semplice: |

|
Post n°325 pubblicato il 24 Luglio 2015 da paoloalbert
Era da qualche anno che mi ero segnata come "da fare" la sintesi dell'acido citrazinico, questo bell'eterociclico che si vede qui accanto. Avete presente il canto delle sirene che ammaliava i marinai?
Fase iniziale, dopo 15 min Fase finale, a 155°
Ottenere "un caramello denso e nero" è di per sè un pessimo indizio durante una sintesi, ma tant'è, la procedura è questa e non si scappa. Tentiamo di andare avanti con fiducia.
Acidificando ora (ottimisticamente con AcOH, ma meglio con acido cloridrico) il sale sodico, DOVREBBE precipitare una polvere gialla (da purificarsi in seguito) di acido citrazinico, il quale è insolubile in acqua.
(Sarei anche curioso di sapere cos'è esattamente questo fango nero rimasto sul filtro, ma sarebbe chiedere troppo)
Il tristissimo residuo nero misterioso
|





|
Post n°324 pubblicato il 17 Luglio 2015 da paoloalbert
C'è stato un periodo a cavallo tra gli anni quaranta e cinquanta dello scorso secolo in cui impazzavano certi ingenuissimi film di fantascienza con razzi a forma di supposta e dischi volanti fatti esattamente come due piatti da minestra sovrapposti.
Si continua l'aggiunta scaldando cautamente e regolando l'aggiunta di (poco) HNO3 in più porzioni fin che tutto il selenio sia ossidato, ovvero fino alla scomparsa dei granelli scuri.
Nel mio caso il colore è leggermente rosato perchè sono partito da un selenio di autoproduzione, non perfettamente puro. |



|
Post n°323 pubblicato il 07 Luglio 2015 da paoloalbert
Contro l'afa che si taglia con un coltello e i trentacinque gradi delle pianure, ho avuto la fortuna di gustare per un po' il fresco degli otto (!) gradi e l'aria salutare della vecchia miniera di rame di Predoi in Val Aurina.
|



|
Post n°322 pubblicato il 01 Luglio 2015 da paoloalbert
Circa una settimana dopo, 30 giugno, sempre alla stessa ora: occhi al cielo.
La Luna, discreta e fuori campo verso sud, se ne sta anche lei a guardare la congiunzione tra Zeus e Afrodite. |

|
Post n°321 pubblicato il 21 Giugno 2015 da paoloalbert
Avete visto che meraviglioso triangolo appare tutte le sere in questo periodo?
|

|
Post n°320 pubblicato il 06 Giugno 2015 da paoloalbert
|

|
Post n°319 pubblicato il 19 Maggio 2015 da paoloalbert
- Guarda questo quadro... io ci vedo il CW nella sua massima espressione! Tu cosa ci vedi? |

|
Post n°318 pubblicato il 11 Maggio 2015 da paoloalbert
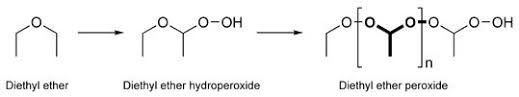
Tutti quelli che hanno a che fare con la chimica sanno che nell'etere possono formarsi delle sostanze che godono di un aggettivo magico: "esplosivo!".
Naturalmente le quantità sono minime, pochi milligrammi per litro, e di nessun pericolo fin che le sostanze sono in soluzione.
La probabilità che mi esploda la bottiglia mentre la apro la ritengo pari più o meno a zero. Segno evidente che il pericolo, in ambito di normale laboratorio, è molto più teorico che reale. |

|
Post n°317 pubblicato il 01 Maggio 2015 da paoloalbert
I miei "Molinari" sono rispettivamente del 1918 (Inorganica) e del 1927 (Organica).
I "Molinari" sono dei libroni che si leggono pian piano e con piacere, oggi più di ieri, senza mai dare al lettore quel senso spiacevole di "mi tocca studiare" che danno certi profondissimi ma gelidissimi tomi attuali.
|


|
Post n°316 pubblicato il 18 Aprile 2015 da paoloalbert
Il tenente Giovanni Drogo aspettò i Tartari invano.
A forza di tartari, mi è venuta voglia di rileggere, per l'ennesima volta, quel capolavoro di cui dicevo all'inizio. |


|
Post n°315 pubblicato il 13 Aprile 2015 da paoloalbert
La 740 è una famosa locomotiva, costruita nel periodo a cavallo della prima Guerra Mondiale, anni prima, anni dopo. Mezza storia dell'ingegno umano passa certamente tra questi due estremi.
La 740 si gira a Ponte nelle Alpi
Rifornimento d'acqua e spalatura carbone a Feltre
Interno di un "Centoporte"
"Recluta" in attesa dell'imbarco per la battaglia del Piave... |




|
Post n°314 pubblicato il 05 Aprile 2015 da paoloalbert
Ogni tanto viene il turno di qualcosina di elettronico ed è quello che capita oggi. In Arduino UNO rimangono a disposizione come IN/OUT solo i pin 0,1,2,3,12,13,A5, sufficienti per farci qualcos'altro.
|

Inviato da: paoloalbert
il 02/05/2021 alle 21:53
Inviato da: Bepi1249
il 15/04/2021 alle 14:11
Inviato da: Tanner85
il 06/02/2021 alle 00:01
Inviato da: poetryclub
il 04/10/2020 alle 23:00
Inviato da: poetryclub
il 04/10/2020 alle 20:58