CHIMICA sperimentale
Esperienze in home-lab: considerazioni di chimica sperimentale e altro
Messaggi di Ottobre 2011
|
Post n°139 pubblicato il 30 Ottobre 2011 da paoloalbert
No, purtroppo le femmine non c'entrano con il discorso che andrò a fare.
ecco come si presenta la valvola 6E5 e a fianco la vista della sommità illuminata, con ancora un settore d'ombra che si potrebbe restringere o allargare. Appropriato il nome "occhio", vero? Lo zoccolo della valvola è di tipo vecchio (anni '30) a 6 piedini, ma ne esiste anche una versione più moderna (anni '40) a otto piedini (in questo caso è detto "octal"). - pin 1: non collegato
|


|
Post n°138 pubblicato il 27 Ottobre 2011 da paoloalbert
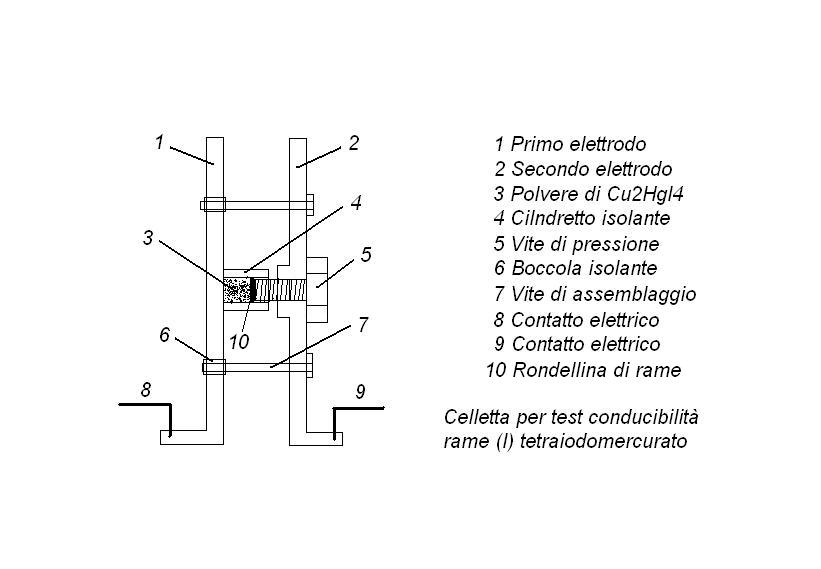
Nella discussione dedicata al termocromatismo del tetraiodomercurato rameoso Cu2HgI4 mi ero riproposto di allestire un setup adeguato alla misura delle variazioni della conducibilità elettrica relativa di questo interessante sale al variare della temperatura. Pertanto occorre pensare e assemblare un dispositivo che possa permettere una notevole compressione meccanica della sostanza in esame e contemporaneamente stabilire i relativi contatti elettrici senza incertezze.
Le immagini e il disegno in sezione spiegano la costruzione di questa celletta dedicata al Cu2HgI4.
Gli elettrodi sono ricavati da due profilati in alluminio a L, serrati assieme da quattro viti di tiraggio, isolate da una parte per mezzo di boccoline in nylon (quelle usate per l'isolamento dei transistor in TO3) in modo che le viti non costituiscano contatto elettrico tra le due piastrine.
Mi aspettavo uno scalino di transizione molto più netto intorno ai 70°, mentre ho notato che le variazioni sono macroscopiche ma progressive. Sottolineo fortemente che tutte queste misure sono RELATIVE, in funzione stretta delle condizioni di lavoro alle quali io ho operato, ma che comunque rendono bene l'idea che mi ero proposto, cioè di evidenziare "le variazioni" e non i valori assoluti, che in questo caso non sono di alcun interesse. Per curiosità ho testato anche il comportamento in media frequenza (fino a 100 KHz) di questa sostanza, senza alcun risultato significativo in semiconduttività: solo pura resistenza ohmica. Per la spiegazione del fenomeno rimando a quanto detto nel post precedente, trovando facilmente in rete ragione del fatto che ad un certo punto con l'aumento della temperatura gli ioni rame si mettono a saltellare di qua e di là nel reticolo cristallino in cerca di "buche", e così facendo (ricordo che --> cariche elettriche che si spostano = corrente!) rendono questa sostanza conduttiva con queste caratteristiche. |




|
Post n°137 pubblicato il 22 Ottobre 2011 da paoloalbert
Il Carnevale della chimica di fine ottobre, ospitato dal blog di Popinga, ci porta in tavola un argomento sfizioso: "La chimica in cucina", lasciando come il solito ai partecipanti una trattazione del tema molto discrezionale. Per non appesantire il discorso ometto i calcoli per risalire al numero di iodio del mio olio, che comunque mi ha dato un valore di 88, in linea con i valori dell'olio di oliva, che possono variare a seconda del tipo e della provenienza tra 80 e 88. (Nel mio caso si trattava di un olio della riviera gardesana, che detto per inciso è un'eccellenza...). |
|
Post n°136 pubblicato il 18 Ottobre 2011 da paoloalbert
Alcune sostanze si comportano in modo strano a seconda delle condizioni a cui sono sottoposte: c'è chi mostra fluorescenza, chi fosforescenza, chi piezoelettricità, chi piezoluminescenza, chi termoconduttività... ecc.... e c'è chi mostra il fenomeno del termocromatismo (o termocromismo), cioè il colore mostrato dalla sostanza è funzione della temperatura.
Le caratteristiche di questo interessante composto iodo-cupro-mercurico non finiscono qui: come dicevo all'inizio, esso al variare della temperatura mostra anche una notevole variazione di conduttività elettrica, che ho voluto investigare quantitativamente nei limiti dei miei mezzi, costruendo un piccolo dispositivo espressamente dedicato a questa prova.
|


|
Post n°135 pubblicato il 14 Ottobre 2011 da paoloalbert
Come dico spesso, ogni tanto arriva inevitabile la sintesi di un estere. Andiamo quindi ad apparecchiare la cucina: lo chef richiede oggi poca roba, è un piattino estivo molto semplice e fresco, di sicuro risultato e di buon gradimento... olfattivo. Allora, in barba a quei "chimici teorici" che non volendo sporcarsi le mani paragonano i chimici sperimentali a dei cuochi, mi dichiaro apposta cuoco chimico ruspante e così procedo:
-In un pallone da 250 ml porre 100 ml di acido acetico, 20 ml di metanolo e 4 ml di H2SO4 concentrato. Dati i vicini punti di ebollizione del metanolo (64,7°) e del metilacetato (57°) ho usato un grande eccesso di acido rispetto all'alcool (più di 3:1) in modo da spostare l'equilibrio a destra il più possibile verso una esterificazione abbastanza completa ed aver meno problemi nella separazione finale. Stavolta non ho messo fotografie della sintesi, perchè si assomigliano tutte e hanno poco significato; in ogni caso... la saga degli esteri continua! |
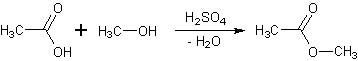
|
Post n°134 pubblicato il 11 Ottobre 2011 da paoloalbert
In occasione del post sui ricordi di scuola ho accennato all'impiego del cannello ferruminatorio; naturalmente si parla a proposito di quegli studenti di chimica che, come me, frequentavano qualche anno fa laboratori dove ancora nessun "apparecchio con la spina" aveva preso piede. I piaccametri li ricordo benissimo perchè, per me che ero stralunato anche nei riguardi dell'elettronica, avevano come indicatore la valvola 6E5GT, uno di quegli "occhi magici" delle antiche radio, con un iride luminescente verde che si apriva e chiudeva secondo l'intensità del segnale, nel caso specifico secondo le indicazioni della sonda al calomelano. L'amico Simone mi ha chiesto come si usavano: ecco come. Le riduzioni sul carbone al cannello ferruminatorio facevano parte dei cosiddetti "saggi preliminari" di una analisi inorganica. Ognuno di noi aveva a disposizione (comprandolo, è chiaro!) un bel blocchetto di carbone di legno di tiglio, una specie di mattoncino resistente lungo un palmo. Il cannello ferruminatorio non era altro che un tubicino di ottone lungo una trentina di centimetri, rastremato in più sezioni e con la punta piegata a L; soffiando nella parte di maggior diametro, si poteva dirigere il dardo concentrato dove si voleva. Essendo il carbone poroso, le sostanze facilmente fusibili come gli alcali sono assorbite, e le altre trasformate prima in carbonati, poi in ossidi e per ulteriore riduzione in metallo. Metalli volatili come Zn, Cd, As, si ossidano comunque e danno un'aureola caratteristica nella direzione opposta a cui si soffia: gialla a caldo e bianca a freddo per lo zinco, marrone per il cadmio, bianca e volatile per l'arsenico (oltre all'odore agliaceo). Si possono riconoscere al carbone anche alcuni sali, per esempio i nitriti, nitrati, clorati, che riscaldati producono microscopiche "deflagrazioni" e certi altri (NaCl per es.) che producono "crepitazione". Naturalmente dopo un po' di tempo il mattoncino di tiglio era tutto bruciacchiato e pieno di aloni e residui di ogni tipo e andava cambiato... ma nel frattempo l'anno scolastico volgeva al terzo trimestre e si passava magari a qualcosa di più "tecnologico", magari andando nel divertentissimo (almeno per me!) Laboratorio di Chimica Organica, al quale ancora oggi assegno con onore le iniziali maiuscole. Tutto molto empirico vero? Certo, molto empirico se vogliamo, ma anche molto divertente!
|
|
Post n°133 pubblicato il 08 Ottobre 2011 da paoloalbert
|

|
Post n°132 pubblicato il 07 Ottobre 2011 da paoloalbert
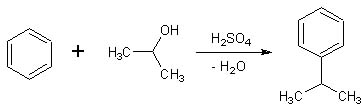
Dando un'occhiata alla mia piccola biblioteca virtuale di chimica organica preparativa mi è capitato di sfogliare un libro edito nel 1972 nella DDR, l'ex Repubblica "democratica" tedesca.
Seguendo e integrando la striminzita procedura del Weygand, ho proceduto in questo modo: Agitare bene e porre a riflusso per tre ore a 65°- In realtà a questa temperatura non bolle niente, si potrebbe forse evitare il refrigerante a ricadere. Morale 2: niente è del tutto sprecato, una esperienza in più rimane comunque ben salda. |
Inviato da: paoloalbert
il 02/05/2021 alle 21:53
Inviato da: Bepi1249
il 15/04/2021 alle 14:11
Inviato da: Tanner85
il 06/02/2021 alle 00:01
Inviato da: poetryclub
il 04/10/2020 alle 23:00
Inviato da: poetryclub
il 04/10/2020 alle 20:58