CHIMICA sperimentale
Esperienze in home-lab: considerazioni di chimica sperimentale e altro
Messaggi di Novembre 2011
|
Post n°147 pubblicato il 29 Novembre 2011 da paoloalbert
L'antefatto
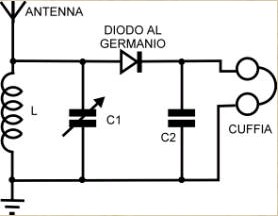
Dove si legge "diodo al germanio" si sostituisca il componente (che allora non esisteva) con l'originale "cristallo di galena" (PbS). Quella minimissima energia elettromagnetica che arriva... diciamo da Londra, entra nell'antenna (un filo lungo almeno una ventina di metri teso più in alto possibile), percorre la bobina L e si scarica a terra (un paletto conficcato in zona umida). Il circuito formato dalla bobina L e dal condensatore C in parallelo, diventa "risonante" su una determinata frequenza (quella della stazione che si intende ascoltare) quando il valori di L e di C sono opportunamente dimensionati, e per questo la capacità del condensatore è variabile con una manopola. In questo modo la componente in alta frequenza del segnale viene eliminata e rimane dopo il diodo solo la bassa frequenza audio derivata dalla modulazione del segnale (qui devo fare un piccolo ma doveroso omissis, altrimenti non ce la caviamo più!). Ma non ho ancora detto perchè si chiama radio a galena. Per tener fede allo spirito pratico-sperimentale del blog, concluderò queste riflessioni con qualche foto della "mia" radio a galena; purtroppo non è quella originale di mio padre, la quale, poverina, mai sarebbe potuta sopravvivere indenne ai massacranti "esperimenti" del giovane sottoscritto... |
|
Post n°146 pubblicato il 24 Novembre 2011 da paoloalbert
Allora oggi dobbiamo bromurare l'alcol amilico.
Materiali occorrenti
Porre a leggero riflusso per 2-3 ore, eventualmente con l'apparato per l'assorbimento acido come si vede in foto; ho notato però che praticamente quasi nulla sfugge dalla bocca del refrigerante. L'alchilbromuro si separa sopra la miscela acida e può essere separato facilmente.
Il bromuro di n-amile si presenta come un liquido limpido pesante (d. 1,22, p.e. 130°), di odore etereo pesante ma piacevole.
Ora il nostro alogenuro è pronto per la futura sintesi di un olezzantissimo tiolo! Quando si parla di tioli il problema "ambiente" si fa arduo, nel senso che è assolutamente improponibile fare queste preparazioni in laboratorio (se lo stesso non è munito di una efficientissima cappa!) perchè l'odore di questi composti col gruppo -SH (l'amilmercaptano è prorio uno di quelli giusti...) è davvero insopportabile ma soprattutto è molto persistente e dove si attacca rimane, vestiti compresi. |
 In pallone da 100 ml introdurre
In pallone da 100 ml introdurre 
 Predisporre quindi il refrigerante Liebig e distillare fin quasi a secchezza il grezzo preventivamente preparato, raccogliendo tra 127-132° (praticamente passa tutto in questo range).
Predisporre quindi il refrigerante Liebig e distillare fin quasi a secchezza il grezzo preventivamente preparato, raccogliendo tra 127-132° (praticamente passa tutto in questo range).
|
Post n°145 pubblicato il 21 Novembre 2011 da paoloalbert
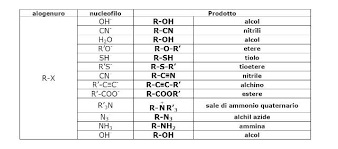
Gli alogenuri alchilici, di formula generale R-X (R = radicale alchilico, X = Cl, Br, I) sono dei reagenti fondamentali per la chimica organica; oltre agli importantissimi reattivi di Grignard possono dare una bella serie di sostituzioni nucleofile, che avvengono naturalmente ognuna nelle condizioni adatte: Nella seconda parte dirò della preparazione pratica del bromuro di n-amile. |
|
Post n°144 pubblicato il 18 Novembre 2011 da paoloalbert
Sembra strano, ma nella vecchia quanto ufficiosa nomenclatura chimica esistevano pure gli alberi, per lo più dedicati all'antica mitologia: e così c'è l'albero di Diana, l'albero di Marte, quello della Luna... e quello di Saturno.
In alto si vede parte dell'anodo e sotto il filo catodico ricoperto dall'alberello ad aghetti e squamette del metallo saturnino che sta crescendo.
In mancanza, ci metto questo elettroalberello. |


|
Post n°143 pubblicato il 14 Novembre 2011 da paoloalbert
Una volta (non l'anno scorso, un po' di più...) bastava andare dallo speziale, dire di essere perseguitati dai topi, chiedere una bustina di "arsenico" ed il gioco era fatto: ecco una decina di grammi di bianca anidride arseniosa a disposizione per qualsiasi evenienza! Ma poi, nel 1836, a dare una mazzata alle avvelenatrici (come si sa l'uso del veleno è da sempre prevalente appannaggio del gentil sesso!) è arrivato l'apparecchio di James Marsh...
Ecco dettagliatamente in cosa consiste e come veniva adoperato questo geniale congegno per la ricerca dell'arsenico, in uso dalla sua invenzione e fino ad una cinquantina di anni fa o anche meno. |

|
Post n°142 pubblicato il 11 Novembre 2011 da paoloalbert
Noi siamo ormai avvezzi per sovradosaggio mediatico ad ogni sorta di notizie di cronaca nera, tanto che non ci facciamo quasi più caso (se non nell'immediato), come drogati che devono aumentare sempre la dose di sostanza per risentirne gli effetti, i quali ben presto svaniscono. |
|
Post n°141 pubblicato il 08 Novembre 2011 da paoloalbert
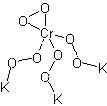
Tutti sanno che il cromo prende il nome dal variegato colore dei suoi composti nei diversi stati di ossidazione.
La reazione che faremo avvenire è la seguente:
|

|
Post n°140 pubblicato il 03 Novembre 2011 da paoloalbert
Ho ritrovato il mio vecchio cannello ferruminatorio di quand'ero studente.
Prima di tutto, quando si rientra nella civiltà dopo lunghi viaggi in paesi remoti, occorre un bel bagno ristoratore; nel suo caso c'è voluto non un bagno ristoratore ma uno "restauratore", a base di... cartavetrata.
Bentornato fra i tuoi amici, cannello! |




Inviato da: paoloalbert
il 02/05/2021 alle 21:53
Inviato da: Bepi1249
il 15/04/2021 alle 14:11
Inviato da: Tanner85
il 06/02/2021 alle 00:01
Inviato da: poetryclub
il 04/10/2020 alle 23:00
Inviato da: poetryclub
il 04/10/2020 alle 20:58