CHIMICA sperimentale
Esperienze in home-lab: considerazioni di chimica sperimentale e altro
|
Post n°373 pubblicato il 12 Aprile 2017 da paoloalbert
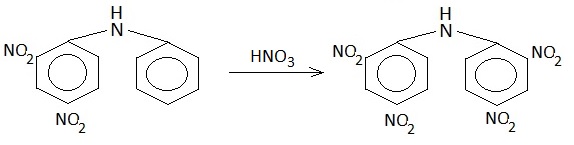
Ed eccoci al successivo intermedio della serie in programma, che è partita dal clorobenzene per arrivare a...
Dopo alcune aggiunte l'agitazione magnetica non basta più perchè la miscela diventa via via più viscosa ed occorre aiutare l'ancoretta magnetica mescolando la pastella anche a mano, magari con l'aiuto del solito termometro maneggiato cautamente e che ci permette di tenere la temperatura sotto perfetto controllo.
Ho ottenuto 9,5 g di 2,2',4,4'-tetranitrodifenilammina, con una buona resa dell'88%. |



|
Post n°372 pubblicato il 03 Aprile 2017 da paoloalbert
L'undici aprile 2017 ricorrerà il trentesimo anniversario della morte di Primo Levi, chimico sperimentale, letterato e storico (tutti e tre al medesimo livello) del quale io ho infinita ammirazione. |
|
Post n°371 pubblicato il 21 Marzo 2017 da paoloalbert
Qualche tempo fa, parlando dell'acido borico (post n. 365) dicevo alla fine che avrei sperimentato un inconsueto metodo di ricerca analitica di quest'acido, ma che l'avrei potuto fare solo in primavera; avrei dovuto dire più precisamente: "DOPO LA FESTA DELLE DONNE".
Per farla breve: passato l'otto marzo e svanita sia la festa che i fiori di mimosa, ho preparato un estratto alcolico dei medesimi e qualche soluzione a diluizione crescente (1 g/l e 100 mg/l e altre di acido borico H3BO3) ed ho eseguito il test di Robin come citato dal Molinari.
A secco, dopo evaporazione, i pozzetti sembrano quasi uguali
Dopo l'aggiunta di ammoniaca. Da sinistra a destra: prova in bianco, 100 mg/l, 1 g/, 2 g/l di H3BO3 - Il colore rosso non appare nel pozzetto di sinistra ma è arancio, la foto è poco fedele. Anche con quantità inferiori a 100 mg/l la colorazione rossa è evidente in presenza di boro. Ho provato anche a fare molti dei test "a umido", cioè senza la noia dell'evaporazione; la reazione avviene comunque ma è meno sensibile.
La formula della curcumina, con tutti quei doppi legami coniugati (deve essere un colorante per forza!) si deduce dalla rosocianina qui sopra; della formula della "mimosina" (nome di fantasia inventato sui due piedi) non ho finora notizia documentata, se non che il metodo funziona, che mi sono divertito a farlo e che lo dedico alle donne (così poco apprezzate in qualche altro ambiente "chimico") che casualmente e pazientemente leggessero fin qui. |





|
Post n°370 pubblicato il 10 Marzo 2017 da paoloalbert
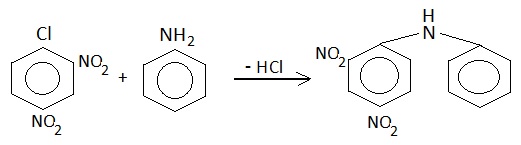
Tempo fa, nel post n. 245, avevo preparato una quantità maggiore del solito (ca. 30 g) di 2,4-dinitroclorobenzene perchè avevo già allora l'idea di utilizzarlo come futuro intermedio... di un intermedio... di un intermedio... eccetera, fino ad arrivare...
[*evidenzio anche in questo caso il mezzo di riscaldamento anacronisticamente adottato: il buon vecchio bunsen e una orrenda reticella decisamente da cambiare. Cose dell'altro mondo rispetto ai lab serissimi? Può darsi... però basta starci attenti e anche in uno sgangherato lab come il mio si riesce a fare con rustici mezzi (proprio come si faceva un tempo!) quasi quel che si vuole...*].
E' una sintesi veramente di soddisfazione visiva come poche, perchè essendo il prodotto molto leggero, sembra tantissimo; la resa è stata di 16,5 g, pari all'82%. |


|
Post n°369 pubblicato il 03 Marzo 2017 da paoloalbert
Una volta i famosi Intervalli TV erano costituiti da pecorelle al pascolo e i sottofondi sonori più trasgressivi erano a base di arpa... pling...plong...pling... una nota ogni quarto d'ora. Ora questo carbonato è facilmente trasformabile in qualunque altro sale di bario e quindi almeno il catione non è stato minimamente sprecato nella sintesi precedente. -Nulla si crea, nulla si distrugge, tutto si trasforma!- predicava quella buon'anima di Antoine-Laurent prima che gli tagliassero la testa. |
|
Post n°368 pubblicato il 24 Febbraio 2017 da paoloalbert
Ahi, ahi, che titolo pretenzioso per una ciambellina senza buco! |
|
Post n°367 pubblicato il 10 Febbraio 2017 da paoloalbert
Mi ero fatto fare da un artigiano ceramista, tempo fa, un bel bicchierino di materiale poroso da usare in esperienze di elettrochimica.
Testo ed immagini, non c'è bisogno di sottolinearlo, sono anni luce lontane dal nostro tempo e proprio per questo suggestive.
|





|
Post n°366 pubblicato il 21 Gennaio 2017 da paoloalbert
In Inorganic Syntheses mi sono imbattuto nella preparazione di un sale che avevo già provato a fare qualche anno fa, basandomi su un'altra fonte e con esiti allora abbastanza infelici.
La resa è stata scarsina come previsto, 4,5 g pari a circa il 67%, ma stavolta il risultato è stato indubbiamente migliore. |

|
Post n°365 pubblicato il 08 Gennaio 2017 da paoloalbert
Passando dalla zona delle Colline Metallifere toscane, dopo San Galgano ed il suo ferro trovato per caso (post n. 361), ho voluto vedere l'originale acido borico di Larderello.
- Nelle Maremme toscane e specialmente a Larderello e paesi vicini, da alcuni crepacci del terreno escono dei soffioni di vapor d'acqua molto caldi chiamati anche fumaroli, che contengono una piccola dose di acido borico insieme a CO2, NH3, H2S e alquanto solfato ammonico. Così brevemente il Molinari a proposito di questo simpatico acido; ricordo, a conferma di quanto afferma il grande Ettore, che la mia vecchia nonna teneva sempre nel suo armadio una bottiglia di "acqua borica" (una soluzione al 3% di H3BO3), che usava appunto come blando disinfettante oftalmico. Per la ricerca analitica dell'acido borico esiste anche un singolare metodo che voglio provare, ma la prova la posso fare solo in primavera (a suo tempo si scoprirà perchè). |

|
Post n°364 pubblicato il 21 Dicembre 2016 da paoloalbert
Oggi è il solstizio d'inverno, il sole splende e da domani splenderà un pelino più a lungo!
Sarebbe il tempo ideale per delle magnifiche diazotazioni "on the rocks" (la materia prima per tenere ghiacciato l'ambiente di reazione non mancherebbe!) ma mancano i reagenti, quelli inediti, bellissimi, interessanti e costosi, che ho deciso di non rinnovare. |

|
Post n°363 pubblicato il 12 Dicembre 2016 da paoloalbert
Ovvero la NON-piramide magica che suona un concerto mirabile di corrispondenze numeriche, cosmiche ed esoteriche.
La casetta magica esoterica con alcuni attrezzi divinatori: Vanga, Forca, Forcone. |

|
Post n°362 pubblicato il 18 Novembre 2016 da paoloalbert
Ogni anno ha le sue ricorrenze, anche più di una. |
|
Post n°361 pubblicato il 07 Novembre 2016 da paoloalbert
Qualche giorno fa, in una anomala giornata di fine ottobre quasi estiva, camminando lungo il sentierino che dall'abbazia di San Galgano conduce a Montesiepi mi è capitato casualmente di buttar l'occhio su un sasso che, avendo un po' di esperienza in queste cose, proprio un sasso-sasso non mi pareva.
|


|
Post n°360 pubblicato il 20 Ottobre 2016 da paoloalbert
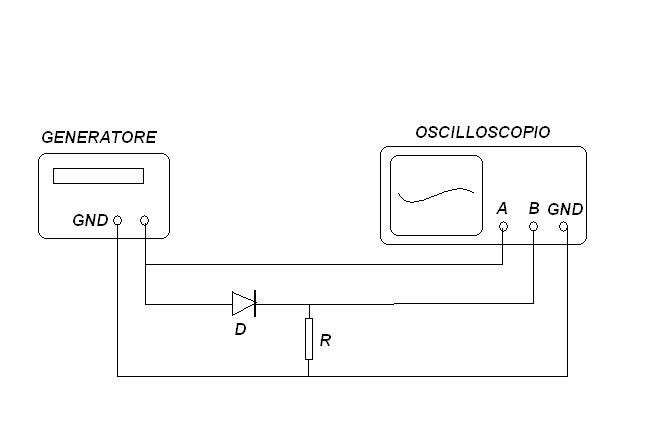
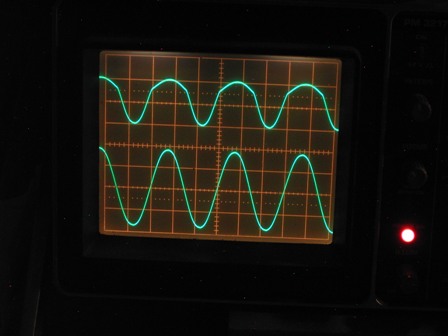
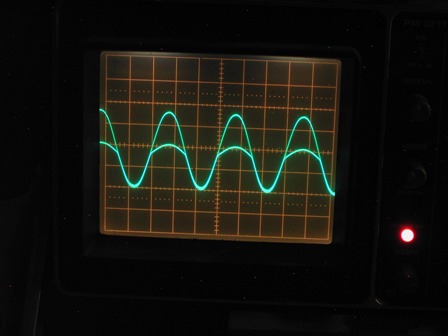
Quale tempo migliore di una uggiosa e piovosa giornata di ottobre per fare un test sperimentale sulle proprietà semiconduttrici del solfuro di piombo cristallizzato, cioè della galena?
Il setup sperimentale, con i contatti sul cristallone di PbS
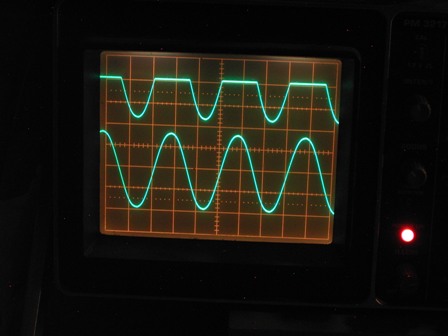
Nella traccia del canale B (in alto) si vede una sinusoide asimmetrica, ben lontano dalla forma ideale vista prima, ma con le semionde positive abbastanza attenuate, segno evidente di un comportamento non lineare del cristallo.
Consideriamo tuttavia che questi rivelatori erano in uso quando i diodi al germanio (e più tardi quelli al silicio) erano ancora di là a venire e nessuno ancora parlava di elettroni e lacune, di drogaggio P e drogaggio N, quindi accontentiamoci abbondantemente. Ricordo che quando da bambino giocavo con la radio a galena era effettivamente assai laborioso trovare il punto sensibile sul cristallo, ma ormai i miei tempi erano maturi per trovare facilmente in commercio i comodisssimi diodi al germanio (i famosi 1N34 o gli OA70), che non abbisognavano di nessuna regolazione ed erano estremamente più sensibili della macchinosa galena d'anteguerra.
Il vecchio detector a galena col baffo di gatto regolabile |


|
Post n°359 pubblicato il 06 Ottobre 2016 da paoloalbert
Più di un mese senza un post... mai successo!
|

|
Post n°358 pubblicato il 01 Settembre 2016 da paoloalbert
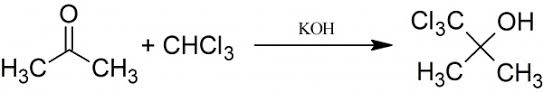
La complessità delle molecole chimico-farmaceutiche moderne spesso, anche se non sempre, è molto elevata; ci sono infatti molecole bellissime e strane, frutto di una ricerca costosa (ma che poi renderà, eccome se renderà!!!, se tutte le sperimentazioni andranno a buon fine).
Si trovano in bibliografia varie procedure, che si differenziano dall'usare come alcalinizzante idrossido di sodio o di potassio, dall'agire in presenza di acqua oppure no, e dall'usare un rapporto reagenti molto variabile. Io ho preferito ripetere quella dell'amico Massimo, che mi dava più affidamento.
|


|
Post n°357 pubblicato il 06 Agosto 2016 da paoloalbert
In questa sede per "chimici sperimentali" intendo coloro che sporcano provette per hobby... diciamo più o meno come il sottoscritto. |
|
Post n°356 pubblicato il 26 Luglio 2016 da paoloalbert
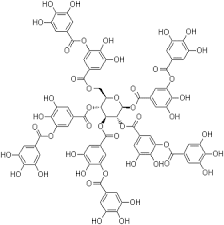
L'acido tannico del commercio si presenta come una polverina amorfa di colore ocra, inodora e di sapore astringente.
Pentadigalloilglucosio - Ac. tannico Acido gallico
Ma abbiamo al nostro arco ancora una freccia da tirare... magari anche due... speriamo che vadano a segno.
[Ho un vecchissimo acido gallico commerciale, di colore quasi identico. Ma in questo caso è dovuto alla veneranda età!]
Lo scopo di questa esperienza era quella di verificare la conversione tannico --> gallico, e mi dichiaro soddisfatto. |




|
Post n°355 pubblicato il 13 Luglio 2016 da paoloalbert
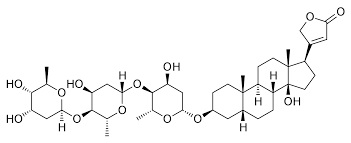
Nel 2004, giacente su un lato della chiesa di Santa Maria Antica alle Arche scaligere di Verona, fu aperto il sarcofago di Cangrande della Scala e riesumato il corpo di quello che fu il signore di quella splendida città fino al 22 luglio 1329, data della sua morte.
Eccola la digitale (Digitalis purpurea), bella e insidiosa, piena di cardiotossici alcaloidi quali i glicosidi digitossina e la digossina, che nelle foglie raggiungono una percentuale non indifferente.
|
 Le analisi effettuate sulla mummia ben conservata sciolsero un dubbio che permaneva da tempo, ovvero se lo Scaligero fosse morto accidentalmente per quella famosa congestione di acqua fredda dopo una cavalcata sotto il sole cocente o se invece fosse stato avvelenato.
Le analisi effettuate sulla mummia ben conservata sciolsero un dubbio che permaneva da tempo, ovvero se lo Scaligero fosse morto accidentalmente per quella famosa congestione di acqua fredda dopo una cavalcata sotto il sole cocente o se invece fosse stato avvelenato.
|
Post n°354 pubblicato il 02 Luglio 2016 da paoloalbert
Finalmente riesco a (ri)sporcare un po' di provette! Ne avevo nostalgia.
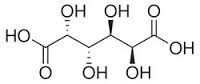
Acido mucico Ossidando vigorosamente un monosaccaride con acido nitrico si produce la trasformazione in due gruppi carbossilici delle funzioni terminali della catena (alcolica e aldeidica) ed un generico acido aldarico ha formula generale HOOC-(CHOH)n-COOH.
Durante il riscaldamento (la T° arriva al massimo a 105°) si ha costante svolgimento di vapori nitrosi ed ipoazotide.
(Dalla soluzione filtrata con opportuna procedura si potrebbe ricavare l'acido saccarico, di identica formula ma che solo per avere un ossidrile che guarda da una parte anzichè dall'altra è solubilissimo in acqua, al contrario del mucico. Potenza della stereochimica!).
Sciogliere il residuo solido nel minimo volume di NaOH diluito (nel mio caso circa 13 ml di NaOH al 10%) e riprecipitare l'acido con HCl diluito fino a netta acidità, sempre oprerando a freddo data che il prodotto ha una leggera solubilità.
|




Inviato da: paoloalbert
il 02/05/2021 alle 21:53
Inviato da: Bepi1249
il 15/04/2021 alle 14:11
Inviato da: Tanner85
il 06/02/2021 alle 00:01
Inviato da: poetryclub
il 04/10/2020 alle 23:00
Inviato da: poetryclub
il 04/10/2020 alle 20:58