CHIMICA sperimentale
Esperienze in home-lab: considerazioni di chimica sperimentale e altro
|
Post n°133 pubblicato il 08 Ottobre 2011 da paoloalbert
|

|
Post n°132 pubblicato il 07 Ottobre 2011 da paoloalbert
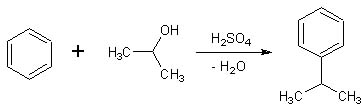
Dando un'occhiata alla mia piccola biblioteca virtuale di chimica organica preparativa mi è capitato di sfogliare un libro edito nel 1972 nella DDR, l'ex Repubblica "democratica" tedesca.
Seguendo e integrando la striminzita procedura del Weygand, ho proceduto in questo modo: Agitare bene e porre a riflusso per tre ore a 65°- In realtà a questa temperatura non bolle niente, si potrebbe forse evitare il refrigerante a ricadere. Morale 2: niente è del tutto sprecato, una esperienza in più rimane comunque ben salda. |
|
Post n°131 pubblicato il 30 Settembre 2011 da paoloalbert
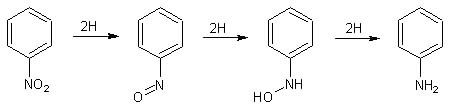
In ambiente acido e fortemente riducente, i nitrocomposti aromatici portano alla formazione della corrispondente ammina; è noto per esempio che il classico metodo di produzione dell'anilina parte dal nitrobenzene che viene ridotto con ferro e acido cloridrico diluito.
la reazione procede dal nitrobenzene, al nitrosobenzene, alla fenilidrossilammina, all'anilina. - In un becker da 250 ml, sciogliere 6 g di NH4Cl in 250 ml di acqua e aggiungere 12 g (10 ml) di nitrobenzene.
L'impiego sicuramente più interessante che se ne potrebbe fare è la sintesi del Cupferron, partendo dalla fen.idr.amm. appena prodotta. Comunque sarebbe un bellissimo reattivo per il rame e per il ferro, ma la sua sintesi presuppone l'impiego di grandi quantità di ammoniaca gassosa proveniente da bombola e quindi tale lavoro è del tutto improponibile per un home-lab. Conclusione: accontentiamoci di vederla per un po', questa fenilidrossilammina, poi una volta nella sua bottiglietta sarà libera di degradarsi a tutto ciò che vorrà, senza alcun rimpianto da parte mia. |


|
Post n°130 pubblicato il 19 Settembre 2011 da paoloalbert
Il tema del Carnevale chimico di settembre, ospitato questa volta da Teresa Celestino sul suo blog Urto Efficace, invita a fare --> |
|
Post n°129 pubblicato il 14 Settembre 2011 da paoloalbert
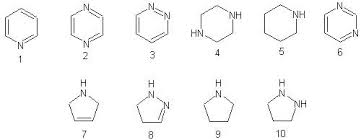
Anche nelle famiglie chimiche succedono cose strane. Ma di cosa stai parlando? Son vaneggiamenti? Niente paura, sto parlando di quell'infornata di sostanze eterocicliche i cui nomi sembrano studiati apposta per far confusione; sfido trovare qualcuno che -tic, tac- sappia sparar fuori tutte le formule giuste, senza incertezze, delle sostanze che dirò. Ricordo intanto per qualche lettore volonteroso che in chimica organica sono cicliche quelle sostanze la cui formula è formata da un poligono, come un serpente che si mangia la coda, ed eterocicliche quelle che hanno intercalato nell'anello della formula anche un elemento diverso dal carbonio. Ecco il campionario delle gemelle PI, spero ci siano tutte le più importanti. Naturalmente faccio questo elenco soprattutto per me, perchè faccio sempre confusione e mai son riuscito (nè mai riuscirò...) ad impararle tutte.
1- Piridina 2- Pirazina 3- Piridazina ... e sotto ci sono quelle con l'anello pentagonale: 7- Pirrolina 8- Pirazolina Le ho messe solo in ordine di "assonanza", niente a che vedere con somiglianze chimiche, aromaticità o quant'altro. - con la pirrolina ci sta il pirrolo... ma ci prova anche il pirrolidone! Visto che ci siamo, vogliamo dare una sbirciatina a qualche appartamento di questo strano condominio, anche se sul campanello il nome non comincia sempre con PI? C'è qualche famigliola moderna, "con gli anelli condensati", e qualche piccola trasgressione... - App.1: l'indolo con l'indolina
Basta! basta! ero partito con l'idea di fare un po' di ordine e mi trovo più confuso di prima! |
|
Post n°128 pubblicato il 10 Settembre 2011 da paoloalbert
Ogni tanto accenno nel blog a quelli che io chiamo "sporcaprovette", cioè a quegli strani individui che hanno come hobby la chimica sperimentale.
Una volta filtrato, possiamo tenere anche questo prodotto bianco cristallino in quantità recuperabile (1 g). Porre il filtrato in una beuta su agitatore magnetico e aggiungere 16 ml di acetone, lasciando mescolare per 4-5 ore per favorire la formazione dell'acetone semicarbazone sotto forma di un precipitato bianco lattiginoso.
Raffreddando in ghiaccio, aggiungere 15 ml di etanolo ed altrettanti di etere, mescolando bene; la semicarbazide cloridato precipita sotto forma di polvere bianca cristallina.
La semicarbazide è un reagente per aldeidi e chetoni perchè forma prodotti di condensazione cristallini con un p.f. ben definito e facilmente separabili, che possono poi rigenerare il prodotto carbonilico per idrolisi acida.
|





|
Post n°127 pubblicato il 06 Settembre 2011 da paoloalbert
Oggi ho perso definitivamente uno dei più fedeli, anzi, il più fedele fra i lettori del mio blog. |
|
Post n°126 pubblicato il 31 Agosto 2011 da paoloalbert
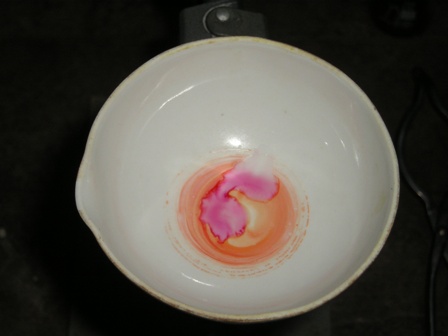
Eccoci finalmente alla fase sperimentale del saggio della muresside: cosa ci serve?
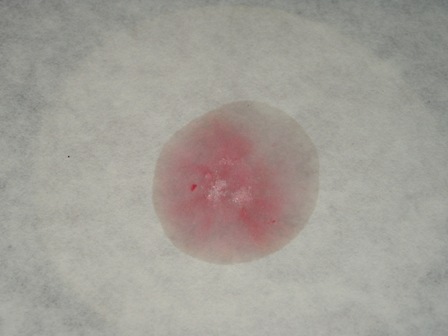
- porre in una capsula una piccola puntina di spatola di caffeina con circa 4-5 volte il suo peso di clorato di sodio o potassio, mescolare e ricoprire la miscela con un paio di ml di HCl concentrato.
Test 2 - porre in una capsula una piccola puntina di spatola di caffeina e aggiungere 1 ml di HCl al 10% e 1 ml di acqua ossigenata al 30%.
Come si vede il test viene benissimo e la colorazione purpurea è molto evidente; chi non avesse a disposizione la caffeina potrebbe estrarne un po' dal tè (anche senza purificarla) e fare un test ancora più "ruspante" sul prodotto alimentare: oso sperare che qualche anonimo e incognito volontario prima o poi lo farà. |




|
Post n°125 pubblicato il 29 Agosto 2011 da paoloalbert
Quando (è passato un bel po' di tempo?) frequentavo con gli amici di scuola il laboratorio di chimica organica, ogni tanto usciva da parte di qualcuno la battuta:
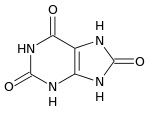
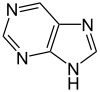
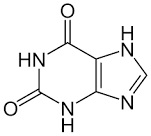
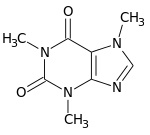
Acido urico Purina Xantina per esempio il derivato trimetilico della xantina è la notissima caffeina del caffè
che con le sorelle quasi gemelle teobromina (del cacao) e teofillina (del tè) costituiscono le cosiddette basi xantiniche.
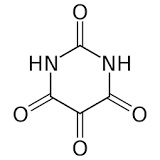
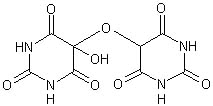
Allossana Acido dialurico i quali possono condensare per formare alloxantina
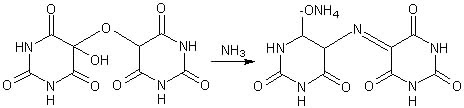
A sua volta l'alloxantina, in presenza di ammoniaca, forma il sale d'ammonio dell'acido purpurico intensamente colorato in rosso purpureo, detto finalmente... muresside!
E' interessante notare che tutti questi nomi pittoreschi dei derivati dell'acido urico vennero scelti per la maggior parte da Wohler e Liebig secondo particolari ragionamenti in un tempo in cui era quasi impossibile intuirne le strutture ed il metodo per determinare il contenuto di azoto era così soggetto ad errori da dare risultati incerti. |

|
Post n°124 pubblicato il 21 Agosto 2011 da paoloalbert
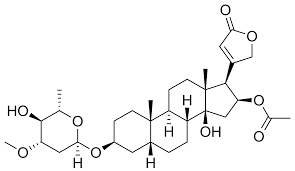
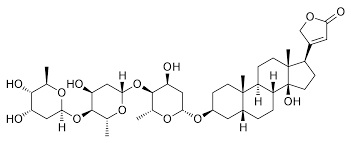
L'ottavo Carnevale della Chimica, ospitato questa volta sul blog Knedliky ha come tema "La chimica delle sostanze bioattive": quale migliore occasione per provare e riferire un paio di test qualitativi che avevo in mente da una montagna di tempo?
Da noi, ed in tutto il Mediterraneo, non c'è persona che non sappia che l'oleandro è una pianta velenosa; certo non dev'essere così dappertutto perchè sembra che specialmente in Sud Africa, dove questa bella pianticella non è autoctona, vi sono molti casi di avvelenamento, soprattutto fra i bambini (la fame dev'essere tanta per riuscire a biascicare queste amarissime foglie...).
L'oleandrina pura si presenta come una polvere bianca cristallina, insolubile in acqua, molto amara. Nella pianta dell'oleandro, specialmente nelle foglie, è contenuta per circa lo 0,08% -
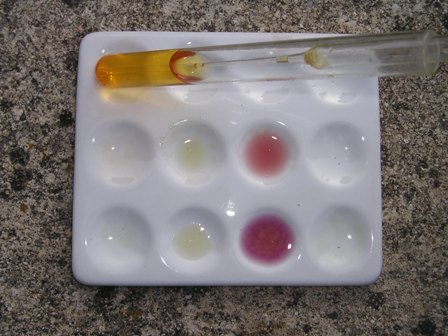
Test n. 1:
Test n. 2
Foto sopra: nel pozzetto a sinistra soluzione diluitissima di oleandrina in etanolo ---°°°OOO°°°--- |


|
Post n°123 pubblicato il 19 Agosto 2011 da paoloalbert
Ecco un altro piccolo intermezzo, facile facile, scaturito giocando d'estate con i sali di cobalto.
sol. di acetato di cobalto complesso con KOCN oppure diluizione con acqua oppure aggiunta di etanolo.
Come dicevo all'inizio, giochetti estivi di poco impegno! |


|
Post n°122 pubblicato il 13 Agosto 2011 da paoloalbert
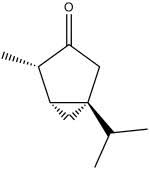
E' incredibile come la mente possa, partendo da uno stimolo qualsiasi, mettersi a girovagare in un milione di agganci, solo con un po', diciamo così, di "predisposizione".
per pura curiosità e a mio rischio e pericolo metto pure il nome ufficiale IUPAC:
Come per ogni sostanza, prima di esprimere spicce considerazioni qualitative, approfondiamo l'argomento sulla tossicità della sostanza medesima dal punto di vista quantitativo (la DL50 e la quantità effettivamente ingerita sono i dati che contano!) e poi traiamone le personali conclusioni.
|


|
Post n°121 pubblicato il 05 Agosto 2011 da paoloalbert
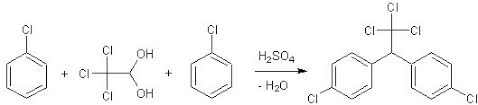
Se tu non fossi proprio giovanissimo e qualcuno ti dicesse: pensa ad un insetticida antimosche, antizanzare, antitutto... pensa ad un classico insetticida da dare con il vecchio spruzzatore a pompetta... tu a cosa penseresti? Tempo di riflessione mezzo secondo: al DDT!
Materiale occorrente: La miscela diventa subito leggermente lattiginosa; mescolare sempre per un'ora circa e poi lasciar riposare fino al giorno successivo, con tappo ermeticamente chiuso. Ricristallizzare il prodotto da alcol isopropilico (80 ml isopropanolo + 20 ml acqua) e lasciar raffreddare lentamente. Filtrare e seccare all'aria.
Resa 3,3 g (circa 46%) di DDT, compresa la discreta quantità dei suoi isomeri (il cloro è in -p, in -o e mix). |
 In una beuta da 100 ml con tappo smerigliato, porre
In una beuta da 100 ml con tappo smerigliato, porre  Mescolare ancora e lasciar riposare ulteriori 4 giorni o anche più, mescolando ogni tanto il liquido giallino torbido.
Mescolare ancora e lasciar riposare ulteriori 4 giorni o anche più, mescolando ogni tanto il liquido giallino torbido. 
|
Post n°120 pubblicato il 30 Luglio 2011 da paoloalbert
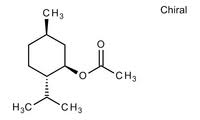
Non c'è niente da fare: ogni tanto il nostro "cuoco" si mette a fare una sana Fischer e poi ci propina l'estere con la scusa di farcene sentire l'olezzo... vabbè, andiamo allora a gustarci questo ennesimo sfizioso piattino: l'acetato di mentile!
Mi sono basato su una procedura eseguita da un bravo sperimentatore sporcaprovette anche lui e siccome questo estere aveva tre buone caratteristiche: 1)- è facile da fare ne ho volentieri sacrificato qualche grammo per sentire la differenza olfattiva tra il mentolo ed il suo derivato acetico.
Sistemare un refrigerante allhin e portare a lento riflusso per un paio di ore. |

|
Post n°119 pubblicato il 24 Luglio 2011 da paoloalbert
Eccoci finalmente alla Miniera di Monteneve, Val Ridanna, Sud Tirol.
Le opere di trasporto del minerale dalla zona di scavo hanno semplicemente dell'incredibile e la mia descrizione non ne renderebbe minimamente giustizia, quindi non la tento nemmeno. L'estensione delle gallerie, che si svolge su una infinità labirintica di livelli, è semplicemente pazzesca (circa 150 Km!), dei quali ovviamente pochissimi sono messi in sicurezza e visitabili, ma più che sufficienti a rendere un'idea esatta delle condizioni di vita e lavoro dei minatori in un posto simile. Ecco quello che sono riuscito a "scavare" dopo una gragnuola di martellate in uno gneiss compatto e durissimo: uno sputo di blenda/galena polverizzato, souvenir che mi son portato via per eseguirvi i test per l'argento, per soddisfare la mia solita curiosità chimica.
Alla fine della giornata, tra le varie considerazioni che si fanno in macchina al ritorno, a tutti noi è venuto spontaneo un interessante paragone sociologico, se così vogliamo chiamarlo. |



|
Post n°118 pubblicato il 21 Luglio 2011 da paoloalbert
Ecco un piccolo post interlocutorio... uno stacchetto insomma. Oggi sul bancone del mio lab batteva uno splendido raggio di sole, il cielo era terso e fuori soffiava un venticello che rendeva la vivibilità perfetta. Mi son detto che nel blog ogni tanto ci vuole un tocco chimico-artistico, magari una bella elio-fluorescenza, vista l'occasione...
Quello che si vede è una provetta bagnata con una soluzione molto diluita (circa 1%) di acido antranilico in glicerina e colpita dal famoso raggio di sole che fa venir subito sera.
Enjoy |


|
Post n°117 pubblicato il 15 Luglio 2011 da paoloalbert
Chimica ed Elettricità, recita il tema del Carnevale della Chimica, ospitato per questa settima edizione sul blog Storie di Scienza di Giovanni Boaga.
Il messaggio, per i pochissimi che non conoscessero il Morse, dice: Come si vede i caratteri sono ben visibili; la nitidezza lascia invece un po' a desiderare, ma la colpa è dei rudimentalissimi attrezzi di scrittura dei quali mi sono servito, visto che non avevo a disposizione una bella macchinetta ottocentesca in ottone. |
 Ho provato a replicare l'esperimento del nostro amico scozzese in maniera semplice ed estemporanea, tenendo ferma la carta e muovendo a mano il pennino:
Ho provato a replicare l'esperimento del nostro amico scozzese in maniera semplice ed estemporanea, tenendo ferma la carta e muovendo a mano il pennino:  Le foto mostrano la base di scrittura, costituita da un foglio di acciaio inox con la carta imbevuta del
Le foto mostrano la base di scrittura, costituita da un foglio di acciaio inox con la carta imbevuta del 
|
Post n°116 pubblicato il 11 Luglio 2011 da paoloalbert
Sfogliando il glorioso e prezioso Treadwell (ai suoi tempi aveva ancora le pagine rilegate in modo da doverle separare col tagliacarte!!!) mi è venuta l'idea di giocare un po' con una vecchia pila zinco-carbone che avevo e di preparare questo "ossido", che rappresenta una buona occasione per studiare un po' la chimica del manganese, non sempre conosciuta come si deve (almeno da me...).
4- Diluire la brodaglia nera con 500 ml di aqua mescolando bene e lasciar decantare pazientemente. Il carbone si separa (un po' galleggia, il resto va in basso).
Quando la soluzione ha riposato quanto basta, pipettare con attenzione il liquido, che deve risultare perfettamente limpido, senza traccia di carbone.
L'ossidazione è solo parziale ed aspettare che coinvolga tutta la massa sarebbe troppo lungo, allora...
9- Il manganito manganoso Mn2O3 (NON è l'ossido del manganese trivalente! E' Mn[MnO3] si presenta come una polvere marrone.
Notare che dopo pochi minuti il colore viola tende a virare a grigio e si ha precipitazione di fosfato di manganese "normale" Mn++ stabile. |
 2- La miscela elettrolitica è costituita da
2- La miscela elettrolitica è costituita da 
 6- Si nota infatti che il precipitato di
6- Si nota infatti che il precipitato di  8- Lasciar sedimentare anche stavolta (anzi più e più volte!) lavando ogni volta con 500 ml di acqua. Questa è la parte molto lunga e noiosa della procedura.
8- Lasciar sedimentare anche stavolta (anzi più e più volte!) lavando ogni volta con 500 ml di acqua. Questa è la parte molto lunga e noiosa della procedura.


|
Post n°115 pubblicato il 30 Giugno 2011 da paoloalbert
Oggi ho deciso di ripassare (queste note le scrivo essenzialmente per me...) la classificazione dei coloranti organici secondo la costituzione chimica, cioè in funzione dei gruppi cromofori presenti nella sua molecola o in funzione della struttura.
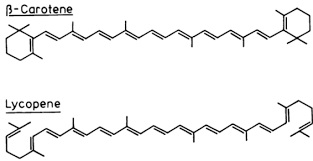
-COLORANTI ETILENICI: contengono il cromoforo etilenico -CH=CH- più volte ripetuto. Importanti in questo gruppo sono i carotenoidi del mondo vegetale: ecco le formule del β-carotene (in alto, giallo) delle carote e del licopene (in basso, rosso) dei pomodori, con la loro bellissima e chilometrica serie di doppi legami coniugati.
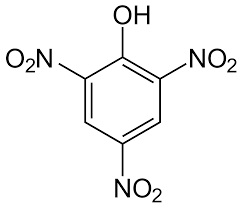
-COLORANTI NITROSO E NITRO: si ottengono nitrosando o nitrando fenoli o naftoli e quindi contengono i gruppi -NO ed -NO2, associati al gruppo ossidrile. Formano facilmente dei complessi di coordinazione con alcuni metalli, generando lacche insolubili che si fissano al substrato. Un esempio è l'α-nitroso-β-naftolo che si colora in rosso legandosi col cobalto, con una sensibile reazione sfruttata in chimica analitica.
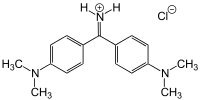
-COLORANTI DEL FIFENILMETANO: hanno il cromoforo chetoimminico C6H5-C(=NH)-C6H5, dove negli anelli aromatici appaiono anche gruppi attivanti in posizione para. Un esempio nei colori classici è il giallo auramina.
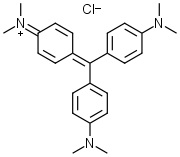
- COLORANTI DEL TRIFENILMETANO: si basano sui gruppi del fucsone (a sinistra) o della fucsoimmina (a destra), dove ai legami liberi del carbonio in alto vengono attaccati altri due anelli aromatici sostituiti in para con gruppi attivanti amminici (più o meno sostituiti) od ossidrili. Esempi noti sono il cristalvioletto ed il verde malachite, metilati nei due gruppi amminici.
-COLORANTI INDIGOIDI: sono caratterizzati dal cromoforo in figura, dove X è un gruppo aromatico più o meno complesso. Naturalmente il colorante più importante di questa classe è l'indaco (naturale o artificiale). Il suo dibromoderivato (due atomi di bromo negli anelli benzenici) costituisce la famosa porpora di Tiro degli antichi.
-COLORANTI XANTENICI: è una classe numerosa in cui è presente la molecola base xantene, poi variamente sostituita e resa più complessa.
-COLORANTI OSSAZINICI E TIOAZINICI: contengono l'anello eterociclico azinico con un atomo di azoto sostituito da uno di ossigeno o di zolfo. Il più rappresentativo è forse il blù di metilene, figura, usato anche in istologia per le proprietà di colorare selettivamente tessuti cellulari.
-COLORANTI FLAVONICI: dulcis in fundo, sostituendo variamente nel 2-fenilcromene (flavone),
alcuni atomi di idrogeno negli anelli benzenici con gruppi alchilici o ossidrilici, si formano molti di quei colori che la natura fornisce ai fiori e ai frutti, ovvero le antocianine.
Al posto di un -R mettiamoci qualche -CH3 o qualche -OH e godiamoci il bellissimo colore risultante, come quello della cianidina, che dà il colore alle rose e ai fiordalsi e modifica il colore a seconda del pH della linfa... Magia della natura, magia della chimica!
L'elenco di cui sopra è sicuramente incompleto, e serve solo come prima veloce individuazione di un colorante organico in una classe definita. Se mi verrà in mente qualcos'altro scartabellando in giro o per qualche buon gradito suggerimento, lo aggiungerò. |
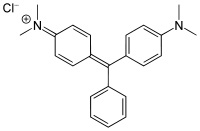
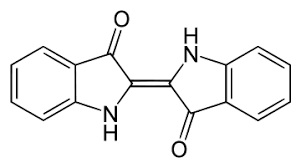
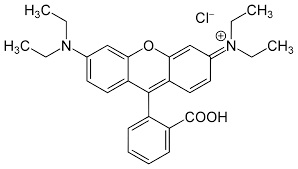
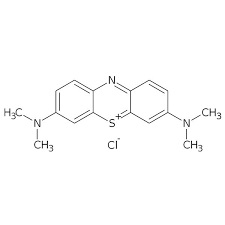
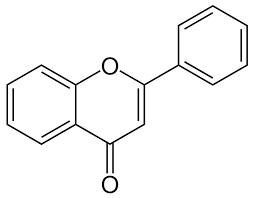
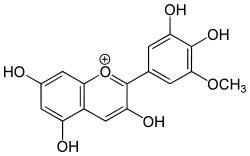
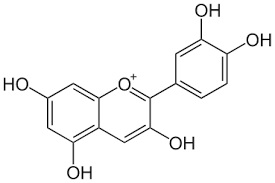 e cambiando qualche ossidrile...
e cambiando qualche ossidrile...

|
Post n°114 pubblicato il 26 Giugno 2011 da paoloalbert
Certi esperimenti costano tempo, denaro e fatica... certi altri non costano proprio nulla...
Ho lasciato semisepolto questo accrocco nella terra per nove mesi, ed ecco i risultati come si vedono in foto. Il rame naturalmente è intatto (a parte una ovvia ossidazione superficialissima) mentre il ferro si è corroso pesantemente, ricoprendosi nei punti non protetti dalla vernice di ruggine profonda (ossido idrato di ferro). Se non fosse stato a contatto del rame si sarebbe ossidato lo stesso, ma in maniera molto più leggera. |

Inviato da: paoloalbert
il 02/05/2021 alle 21:53
Inviato da: Bepi1249
il 15/04/2021 alle 14:11
Inviato da: Tanner85
il 06/02/2021 alle 00:01
Inviato da: poetryclub
il 04/10/2020 alle 23:00
Inviato da: poetryclub
il 04/10/2020 alle 20:58