CHIMICA sperimentale
Esperienze in home-lab: considerazioni di chimica sperimentale e altro
|
Post n°293 pubblicato il 15 Novembre 2014 da paoloalbert
...DELLA NATURA DEL SALNITRO ET DEL MODO CHE A FARLO SI PROCEDE |
|
Post n°292 pubblicato il 10 Novembre 2014 da paoloalbert
...DELLA NATURA DEL SALNITRO ET DEL MODO CHE A FARLO SI PROCEDE |
|
Post n°291 pubblicato il 05 Novembre 2014 da paoloalbert
DELLA NATURA DEL SALNITRO ET DEL MODO CHE A FARLO SI PROCEDE |
|
Post n°290 pubblicato il 01 Novembre 2014 da paoloalbert
Parlavo qualche tempo fa del sal di muro, ovvero di quel ""salnitro"" (le doppie virgolette sono d'obbligo) che riveste talvolta i muri umidi e che salnitro proprio non è (nel mio caso si trattava di Na2SO4, senza traccia nè di nitrati nè di potassio). |
|
Post n°289 pubblicato il 28 Ottobre 2014 da paoloalbert
Ogni volta che apro questo blog, per scriverci o così per fare un giro, è pacifico che mi scappi l'occhio su quelle figurine che stanno a destra, sotto la scritta "Ultime visite al blog". |
|
Post n°288 pubblicato il 15 Ottobre 2014 da paoloalbert
Un volonteroso giovane sperimentatore mi ha chiesto qualche consiglio riguardo la sintesi del formiato di isopropile. |
|
Post n°287 pubblicato il 07 Ottobre 2014 da paoloalbert
Ecco una sintesi facile facile e anche didattica: l'ossidazione diretta degli alcoli ad acidi ad opera del permanganato. Ora viene la parte più noiosa del lavoro, che consiste nel concentrare la soluzione lasciandola evaporare per riscaldamento fino a ridurne il volume a circa una cinquantina di ml.
|
|
Post n°286 pubblicato il 13 Settembre 2014 da paoloalbert
Povero Nikola Tesla, ti hanno demolito la tua Wardencliffe Tower e non sapevano che ti saresti preso una bella la rivincita...
Le notizie che si possono trovare in rete cliccando "tesla" sono ricche e pressochè infinite e quindi non approfondisco; come breve prologo alla realizzazione dirò solo questo: Come generatore di alta tensione ho usato un trasformatore di riga di un vecchio televisore a tubo catodico pilotato da un paio di transistors 2N3055 in un circuito autooscillante; la tensione prodotta a vuoto è di circa 25mila volt (a bassa corrente!) che diminuisce però notevolmente sotto carico.
L'odorino dell'ozono purtroppo non lo posso mettere, cerchiamo di avere pazienza ancora per qualche decennio... |




|
Post n°285 pubblicato il 15 Agosto 2014 da paoloalbert
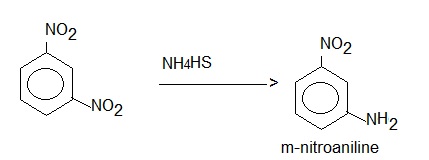
Non è possibile nitrare direttamente l'anilina perchè quest'ultima è troppo sensibile all'ossidazione e l'azione dell'acido nitrico porterebbe solo a prodotti indesiderati (quegli odiosi oliacci neri, incubo di tante reazioni di chimica organica dal dubbio esito...).
Ho provato la sintesi in varie procedure, adattandole opportunamente e cambiando il modo di solfurazione: con sodio solfidrato NaSH, con sodio bisolfuro Na2S2, e con potassio polisolfuro K2Sx ("fegato di zolfo" commerciale). Materiale occorrente - 1,3-dinitrobenzene NO2-C6H4-NO2 -In una beuta da 100 ml sciogliere 5,5 g di sodio solfidrato in 50 ml di metanolo, scaldando un pochino. Raffreddando e filtrando si deve ottenere una soluzione limpida. Versare il residuo in 200 ml di acqua ghiacciata; la m-nitroanilina precipita in fiocchi aranciati e si separa facilmente per filtrazione su buchner. Lavarla bene con poca acqua gelida e porre il solido in un becher con ancora 200 ml di acqua, portando all'ebollizione.
Questa fase è importante perchè permette di eliminare il poco dinitrobenzene non reagito ed impurezze peciose che si raccolgono alla superficie del liquido; si eliminano facilmente con la punta di una spatolina fredda, alla quale aderiscono. Rifinire eventualmente con qualche strisciolina di carta da filtro. Lasciar raffreddare lentamente per avere una buona cristallizzazione (io ho avvolto il becher con un panno e lasciato al decrescente calduccio per qualche ora).
La m-nitroanilina è una delle poche sostanze aromatiche che ha il vantaggio di lasciarsi ben cristallizzare senza dover ricorrere a solventi perchè in acqua è molto solubile a caldo e poco a freddo (circa 0,1 g/100 ml).
Ricordo che questa sostanza è molto tossica ed occorrono quindi tutte le avvertenze durante la sintesi per tenerla a bada... noi da una parte e lei dall'altra.
|


|
Post n°284 pubblicato il 08 Agosto 2014 da paoloalbert
Ogni tanto mi metto a risporcare qualche provetta, per non diradare troppo la parte sperimentale di questo blog.
Terminata l'aggiunta porre il pallone in un bagno d'acqua bollente e riscaldare a ricadere per almeno un'ora, mescolando frequentemente.
Filtrare su buchner separando al meglio la parte liquida acida e aggiungere al residuo ancora 300 ml di acqua, riscaldare fino a fusione del prodotto (sotto i 90°) e neutralizzare con NaHCO3 solido fino a cessare dell'effervescenza e ancora un poco in eccesso.
|





|
Post n°283 pubblicato il 29 Luglio 2014 da paoloalbert
Lessi qualche anno fa un bellissimo saggio sull'antica "coltivazione" del salnitro (nitrato di potassio, KNO3), il componente essenziale per la fabbricazione della vecchia polvere nera da sparo.
Ho voluto farla vedere a qualcuno e chiedere cosa fosse: SALNITRO, hanno risposto tutti senza la minima esitazione (naturalmente nessuno sapeva esattamente cosa significasse chimicamente questa parola). Una grattatina al muro ed ecco un bel campioncino di cristallini leggeri e vaporosi (sembrano acido salicilico) pronti a sacrificarsi per l'analisi.
Ma allora il potassio proprio non cè! Macchè, sembra proprio di no. -Sesto test: con nitrato d'argento. Andiamo a vedere se magari ci sono anche i cloruri. La risposta è chiara: Na2SO4.10H2O, ovvero banale SOLFATO DI SODIO decaidrato. |



|
Post n°282 pubblicato il 14 Luglio 2014 da paoloalbert
I temporali praticamente quotidiani di questo mezzo luglio 2014 mi hanno fatto venire in mente i razzi dell'Anselmo.
E che fine ha fatto il prode Anselmo con la sua credenza piena di TNT? |

|
Post n°281 pubblicato il 30 Giugno 2014 da paoloalbert
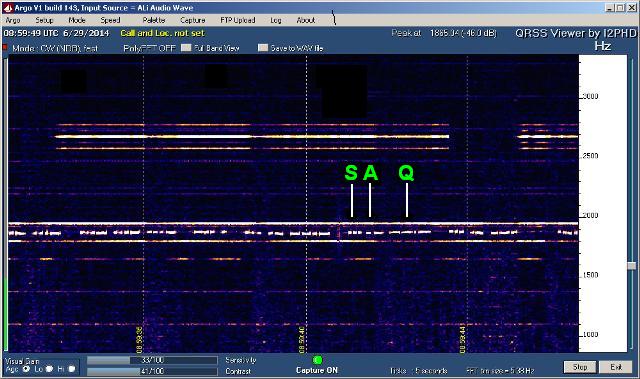
Stavolta di chimica non c'è niente di niente, l'argomento è solo radioricezione; il breve testo che segue è, diciamo così, dedicato a qualche appassionato che per puro caso passasse da queste parti. E' utile fare una sorta di analogia, tanto per capire. La frequenza di ricezione è 17,2 KHz, corrispondente ad una lunghezza d'onda di 17,442 chilometri (chilometri, non metri... se non fosse radiofrequenza saremmo quasi in banda audio!).
L'SPM-15 è sintonizzato...
Lo spettrogramma Argo è poco nitido ma accettabile... |


|
Post n°280 pubblicato il 28 Giugno 2014 da paoloalbert
Ho vicino a casa ancora qualche ciliegio di quelle antiche qualità che mio nonno piantò molti decenni fa; purtroppo molte piante non sono sopravvissute alla terribile estate del 2003, quando il calore tropicale e la siccità fecero strage di alberi da frutto nella mia zona, ma ne rimane tuttavia ancora qualcuna.
Dicono che gli antociani abbiano ottime proprietà antiossidanti e in definitiva facciano bene... beh allora oggi, dopo la raccolta, mi hanno fatto decisamente bene, e "dentro" devo essere colorato come una boccetta d'inchiostro. |


|
Post n°279 pubblicato il 07 Giugno 2014 da paoloalbert
Non mi era mai capitato di vedere in tale quantità questo materiale aderente come una spessa crosta metallica su un pezzo di carburo di calcio, ma solo come piccole inclusioni.
La forma dei frammenti deriva dalla frantumazione di una massa fusa al forno elettrico a oltre 2000° e i pezzi rapidamente imbianchiscono e sfioriscono per reazione con l'umidità atmosferica e vanno conservati in recipienti a tenuta ermetica.
|


|
Post n°278 pubblicato il 31 Maggio 2014 da paoloalbert
C'è stato un periodo nel quale (nella mia zona) tante famiglie contadine avevano in casa una bella latta piena di pezzi di carburo (di calcio, CaC2), che si presenta più o meno come dei "sassi" di forma irregolare.
e questa lampada c'era pure identica (ma non così arrugginita!) anche a casa mia; ma come luce di emergenza, dato che nessuno dei miei famigliari è mai andato a cercar chiocciole. |


|
Post n°277 pubblicato il 24 Maggio 2014 da paoloalbert
Mio padre da giovane (mentre io ero ancora nello spazio cosmico) rischiò quasi di ammazzarsi col carburo.
Dicevo che mio padre se la vide molto brutta: l'incidente successe perchè lo mandarono di sera a controllare la riserva di carburo, e lo mandarono... CON LA CANDELA! |

|
Post n°276 pubblicato il 12 Maggio 2014 da paoloalbert
|
|
Post n°275 pubblicato il 30 Aprile 2014 da paoloalbert
Dopo il rilassante ambiente dell'isola del Minotauro, un po' di chimica. Chi non ricorda (non mi rivolgo ai giovanissimi...) i vecchi esposimetri, detti appunto "al selenio", esibiti trionfalmente dagli appassionati di fotografia di qualche decennio fa? L'esposimetro era la macchinetta che distingeva un vero "fotografo" da un qualsiasi dilettante della domenica. Sempre lui, l'elemento intendo, ebbe anche un breve ed intenso momento di gloria in campo elettronico a cavallo tra gli anni '50 e '60, quando fu molto usato negli apparecchi a valvole quale raddrizzatore per la tensione anodica.
si riusciva a risparmiare due volte: sia una valvola (la raddrizzatrice), sia l'energia per il suo riscaldamento.
La provetta (inquinata con rame) evidenzia la presenza di selenio; la fotografia è vergognosamente povera ma in qualche modo rende l'idea.
H2SeO3 + 2 Zn + 4 HCl --> Se + 2 ZnCl2 + 3 H2O Si pulisce accuratamente una lamina di zinco con carta vetrata, portando al vivo il metallo, e subito vi si pongono un paio di gocce di soluzione di Se4+ in soluzione debolmente cloridrica. Ben presto apparirà una macchia rossa o di color bronzeo, in funzione della concentrazione in Se.
|



|
Post n°274 pubblicato il 17 Aprile 2014 da paoloalbert
|


Inviato da: paoloalbert
il 02/05/2021 alle 21:53
Inviato da: Bepi1249
il 15/04/2021 alle 14:11
Inviato da: Tanner85
il 06/02/2021 alle 00:01
Inviato da: poetryclub
il 04/10/2020 alle 23:00
Inviato da: poetryclub
il 04/10/2020 alle 20:58