
CHIMICA sperimentale
Esperienze in home-lab: considerazioni di chimica sperimentale e altro
Messaggi di Dicembre 2010
|
Post n°69 pubblicato il 30 Dicembre 2010 da paoloalbert
La chemioluminescenza è una caratteristica di pochissime sostanze che in determinate condizioni emettono radiazione nel visibile per un certo tempo.
Al buio totale, dopo aver abituato gli occhi, mescolare 10 ml della soluzione A e 10 ml della soluzione B e porre la miscela in un cilindro; aggiungere in un colpo mescolando i 10 ml della la soluzione C: risulterà una debole ma visibilissima luminescenza giallastra, che perdurerà per qualche minuto!
Ecco finalmente il cigno promesso... un cigno da chimici naturalmente! Enjoy with lophine! |
 A- Sciogliere 1 g di lophine in 20 ml di etanolo tiepido
A- Sciogliere 1 g di lophine in 20 ml di etanolo tiepido

|
Post n°68 pubblicato il 30 Dicembre 2010 da paoloalbert
Come anticipato a Natale e con mia personale soddisfazione devo tirare in ballo ancora una volta il simpatico Aleksandr P. Borodin, il quale cita nella sua prima relazione scientifica (1859) le ricerche effettuate sull'idrobenzamide e sui suoi derivati amarina e 2,4,5-trifenilimidazolo. Procedura La fase seguente va eseguita in modo opportuno poichè vengono emessi vapori tossici ed irritanti.
La prossima volta vedremo come visualizzare la chemioluminescenza della lophine... ancora un po' di pazienza! |

|
Post n°67 pubblicato il 28 Dicembre 2010 da paoloalbert
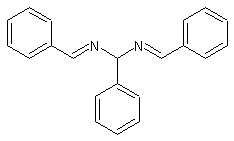
Questo composto (scoperto nel 1836 da A.Laurent, che gli ha dato il nome), è apparentemente una anonima sostanza come migliaia di altre più o meno simili; vedremo invece che l'idrobenzamide possiede una potente chance in più: servì (e servirà a sua volta nella seconda parte di questo lavoro) alla preparazione di un'altra sostanza, decisamente interessante... Ho trovato sul Cumming del 1937 (le fondamenta ciclopiche della chimica sperimentale sono ancorate ai "vecchi" e sacri testi... chi non ricorda il Gattermann di Primo Levi?) una bella sintesina facile facile che ho sperimentato con successo: è una reazione di condensazione e si basa sulla reazione tra l'ammoniaca e la benzaldeide per generare una sostanza che si chiama 1-phenyl-N,N'-bis(phenylmethylidene)methanediamine, e che tutti chiamano molto più amichevolmente hydrobenzamide, oppure, se proprio siamo allergici alla lingua d'oltre Manica, idrobenzamide. Il materiale occorrente è semplice e la sintesi anche, ma già dalla bella formula del prodotto si potrebbe immaginare che esso racchiuda una sorpresina finale; diamo il via dunque alla metamorfosi del brutto anatroccolo, il quale da subito lascia intravvedere con un po' di immaginazione un futuro cigno...
- benzaldeide C6H5-CHO - In una beuta da 100 ml porre semplicemente 10 ml di benzaldeide e 50 ml di ammoniaca concentrata; si forma immediatamente una emulsione bianca; mescolare agitando vigorosamente e ripetere l'operazione ogni tanto per le successive due tre d'ore. L'idrobenzamide si presenta come una polvere bianca (velenosa, classe di rischio T) con odore di benzaldeide (probabile residuo), insolubile in acqua, p.f. teorico 110°, nel mio caso più basso.
Per adesso mettiamo da parte l'anatroccolo; la prossima volta tenteremo di trasformarlo in cigno! Non si vede dalla formula che ha già due bellissime ali? |

|
Post n°66 pubblicato il 22 Dicembre 2010 da paoloalbert
Piccola pausa natalizia dedicata a tutti i visitatori, prendendo spunto dal mio stimato chimico compositore Александр Порфирьевич Бородин (Alieksandr Parfirièvic Baradìn), che presto avrò l'onore di citare ancora una volta riguardo una sintesi che lo toccherà da vicino.
...Buon Natale! |
|
Post n°65 pubblicato il 19 Dicembre 2010 da paoloalbert
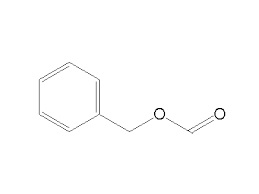
Prima della pausa natalizia voglio completare la trilogia degli esteri benzilici degli acidi grassi inferiori, proponendo la terza e ultima sintesi di un altro estere odoroso importante, quella del formiato.
Materiale occorrente: - In un pallone da 250 ml introdurre 28 ml di alcool benzilico e 46 ml di acido formico all'85%; mescolando aggiungere 2 ml di H2SO4 conc. e predisporre il sistema per riscaldamento a ricadere con mantello riscaldante o con bagno ad olio o sabbia. Ho usato questo sistema, immergendo nella sabbia il termometro. Distillare il prodotto (circa 30 ml) raccogliendo tra 200 e 208°,ottenendo 17 ml di benzile formiato (64%). D. 1,05 - P.e. 203°- Liquido limpido incoloro, oleoso (un po' contro la logica?), con odore fruttato, diciamo una via di mezzo tra il gelsomino e le mandorle, ma più debole e aspro, meno gradevole e diverso dal benzilacetato o propionato.
Sarebbe interessante conoscerne l'effettiva purezza, per verificare la presenza di eventuali prodotti di ossidazione e/o reazioni secondarie... ma ci vorrebbe "un apparecchio con la spina!".
|

|
Post n°64 pubblicato il 12 Dicembre 2010 da paoloalbert
Devo decidermi a mettere ogni tanto qualche intermezzo... in mezzo a queste badilate di chimica, altrimenti si rischia l'indigestione! Spesso quando scrivo qualcosa per questo blog mi ascolto in relax qualche musica preferita; per esempio oggi mi sono ascoltato fra l'altro quel capolavoro di Bach che si trova qui sotto, e che in vicinanza del Natale ci sta a pennello. |
|
Post n°63 pubblicato il 09 Dicembre 2010 da paoloalbert
Oggi faremo un uso della chimica sperimentale molto utilitaristico e pratico; saranno più contenti coloro che di solito non capiscono in queste mie strane riflessioni che gusto ci sia nell'attaccare pezzi di molecola ad un'altra per crearne una terza senza alcun fine pratico, ma solo per farlo così, per pura soddisfazione intellettuale. Oggi, come dicevo, niente di tutto questo! Poichè il mio lab confina in qualche modo anche con la lavanderia di casa, vedendo sullo scaffale una bottiglia di candeggina mi è venuto lo sfizio di verificare in quale percentuale vi fosse contenuto il principio attivo, ovvero l'ipoclorito di sodio NaClO. L'analisi della candeggina (almeno "questa" analisi...) si basa su due reazioni redox; in una reazione redox c'è una sostanza che si ossida e una che si riduce (non sto a dire cosa significa altrimenti non ce la caviamo più...). ClO- + 2 H+ + 2 I- --> Cl- + I2 + H2O Traducendo: l'ipoclorito ossida uno ioduro a iodio e lui si riduce a cloruro. Ecco la seconda: 2 S2O3-- + I2 --> 2 I- + S4O6-- Traducendo: il tiosolfato riduce lo iodio a ioduro e lui si ossida a tetrationato. Poste queste premesse, ho preparato: - una soluzione 0,1 M di tiosolfato di sodio (p.m. 248,10), sciogliendo 2,48 g di Na2S2O3.5H2O in 100 ml di acqua (questa soluzione va fatta esattamente) - una soluzione circa al 5% di acido acetico, diluendo 2,5 ml di CH3COOH in 50 ml di acqua - una soluzione circa al 5% di ioduro di potassio KI, sciogliendone 0,5 g in 10 ml di acqua - una soluzione di amido in acqua, disperdendone 0,1 g in 5 ml di acqua Procedura: - misurare esattamente 10 ml di candeggina e portarli a 100 ml; prendere esattamente 10 ml di questa soluzione e diluirli in un becker con circa 50 ml di acqua - acidificare aggiungendo 10 ml della soluzione di acido acetico - aggiungere circa 5 ml della soluzione di KI; la soluzione assumerà istantaneamente una colorazione marrone, indice che lo ioduro (in eccesso) ha consumato tutto l'ipoclorito ed ha sviluppato la corrispondente esatta quantità di iodio - con una buretta calibrata aggiungere goccia a goccia la soluzione di tiosolfato finchè la colorazione avrà assunto una colorazione giallina (stà diminuendo lo iodio riducendosi a ioduro ed il colore si attenua) - aggiungere qualche goccia della soluzione di amido (la soluzione assume colore violaceo per il complesso che l'amido forma con lo iodio ancora libero) - continuare la titolazione con la buretta, sempre mescolando, finchè la colorazione tende a sparire; il punto di viraggio non è semplice da cogliere, qui è indispensabile un po' di esperienza. Quando il colore tende a sparire vuol dire che non c'è più iodio libero e che tutto il tiosolfato aggiunto si è trasformato in tetrationato. Ora un po' di calcoli... Dall'analisi delle ossidoriduzioni risulta che un equivalente di ipoclorito viene "consumato" da due equivalenti di tiosolfato. Abbiamo fatto una soluzione 0,1 M di Na2S2O3, la quale contiene 15,81 g/l di tiosolfato anidro, ovvero 0,0158 g ogni ml; siccome ad ogni grammo di tiosolfato corrispondono 0,15 g di ipoclorito, ad ogni ml consumato nella titolazione corrispondono quindi 0,0158 x 0,15 = 0,00237 g di NaClO Nel mio caso il punto di viraggio (su tre prove eseguite in sequenza) è stato raggiunto mediamente con 22,5 ml di tiosolfato: 22,5 x 0,00237 = 0,053 Eccoci finalmente al traguardo: Salvo errori ed omissioni, naturalmente! |
|
Post n°62 pubblicato il 05 Dicembre 2010 da paoloalbert
L'altra volta ho parlato in modo discorsivo (e molto incompleto!) degli elementi delle terre rare; oggi l'argomento entra nel particolare della nuda chimica sperimentale e ci entra di brutto. Quanto segue è quindi strettamente riservato agli sporcaprovette impenitenti e amanti degli elementi esotici! Taca banda! |
|
Post n°61 pubblicato il 02 Dicembre 2010 da paoloalbert
Nella splendida biblioteca storica della mia città (non dico quale) ho da poco scoperto che esiste una monumentale opera enciclopedica in francese sulla chimica inorganica che farebbe la felicità di qualsiasi chimico sperimentale. Dopo questa inquietante introduzione, qualcuno (il solito coraggioso...) si chiederà cosa diavolo siano queste Terre Rare, che hanno perfino l'onore della lettera maiuscola. -Scandio, Yttrio |
Inviato da: paoloalbert
il 02/05/2021 alle 21:53
Inviato da: Bepi1249
il 15/04/2021 alle 14:11
Inviato da: Tanner85
il 06/02/2021 alle 00:01
Inviato da: poetryclub
il 04/10/2020 alle 23:00
Inviato da: poetryclub
il 04/10/2020 alle 20:58