CHIMICA sperimentale
Esperienze in home-lab: considerazioni di chimica sperimentale e altro
|
Post n°353 pubblicato il 22 Giugno 2016 da paoloalbert
Sì, missione compiuta, siamo riusciti a camminare sui Christo's Floating Piers! (Riguardo gli avvicinamenti in auto o in battello posso solo immaginare, ma non ho esperienza concreta).
Il Lago d'Iseo verso l'soletta di San Paolo con i floating piers
Altrimenti sarei ancorà là, in fila per il treno di ritorno, con i pompieri che spengono con gli idranti i colpi di calore. |

|
Post n°352 pubblicato il 09 Giugno 2016 da paoloalbert
Aspettando l'occasione di tornare a sporcare un po' di provette (prima o poi l'occasione arriverà, un po' di fiducia...) girovagando in rete in chi m'imbatto?
Ma non sarà "uno dei miei mongoli"?, mi dico. E vado a verificare nelle mie vecchie foto... sì, è lui!
Esattamente 10 anni fa dunque mi trovavo a Parigi, vicino al Beaubourg, ritrovo di tutti gli artisti di strada d'ogni parte del mondo. Con l'occasione mi deliziai ad ascoltare un quartetto musicale mongolo, gli Altai Khairkhan, ed era come avere un pezzo di Mongolia a portata di mano, anzi, di orecchio.
|


|
Post n°351 pubblicato il 22 Maggio 2016 da paoloalbert
Ma dov'è andata a finire la chimica sperimentale di questo blog?
A mia discolpa c'è stato nell'ultimo anno il poco tempo a disposizione da dedicare agli hobbies, spero che la situazione migliori con la prossima bella stagione. |

|
Post n°350 pubblicato il 15 Maggio 2016 da paoloalbert
Se amate la scienza e soprattutto la tecnologia come le amo io, allora DOVRESTE andare, almeno una volta nella vita, a girovagare per i corridoi del Deutches Museum di Monaco.
"Laboratorium zur Zeit Lavoisier"
|


|
Post n°349 pubblicato il 06 Maggio 2016 da paoloalbert
Qualche giorno fa sono andato in un negozio specializzato in ricambi domestici per cercare i manici di una pentola. |
|
Post n°348 pubblicato il 25 Aprile 2016 da paoloalbert
Oggi, 25 aprile, ricorre anche un altro anniversario: quello della nascita di Guglielmo Marconi.
Finalmente, dopo essere riusciti a distruggere e disperdere finanche quel cimelio inestimabile della nave marconiana Elettra (di cui ho già parlato), arriva il decreto legge del 22 dicembre 2008 n.200, che nelle "Misure urgenti in materia di semplificazione normativa" recita sulla Gazzetta Ufficiale che: |

|
Post n°347 pubblicato il 10 Aprile 2016 da paoloalbert
Qualche giorno fa sono passato lungo la strada (pochissimo frequentata) che della valle conduce al monte ed a fianco del solito tornante ho trovato in pieno fiore il prugnolo selvatico che a fine estate mi premia.
Fiori di prugnolo, simili al biancospino
|

|
Post n°346 pubblicato il 17 Marzo 2016 da paoloalbert
[Intermezzo fino al prossimo esperimento] - Certo Teresa, è una di quelle cose che ti fanno venire il cancro! - Ma tu ci stai alla larga? - Magari! Se potessi! Ma siamo tutti bombardati dalle onde elettromagnetiche, dannate multinazionali... @#!gr!#@!&*arrgr! |
|
Post n°345 pubblicato il 08 Marzo 2016 da paoloalbert
Eugenio Bertorelle, classe 1913, fu tre cose insieme: un chimico, un professore ed un artista. Ho preparato la soluzione dei vari sali in 200 ml di acqua e predisposto la celletta elettrolitica.
Con la medesima soluzione effettuerò altre due prove in condizioni diverse e ne parlerò a suo tempo: |



|
Post n°344 pubblicato il 28 Febbraio 2016 da paoloalbert
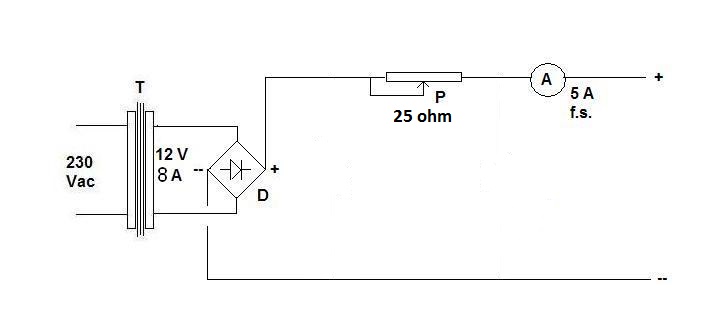
[Galvanotecnica è intesa come il modo di ricoprire elettroliticamente un metallo con un altro]
|
|
Post n°343 pubblicato il 18 Febbraio 2016 da paoloalbert
C'è una sperduta ma interessante località presso il lago Bernic, nel Manitoba (Canada). Questo metallo, il cui nome significa "blù cielo" dal colore delle sue righe spettroscopiche osservate da Bunsen e Kirchoff che ne permisero la scoperta un secolo e mezzo fa, è il CESIO.
Se lo tocchi ti brucia, perchè la pelle contiene acqua e non c'è nulla come questa sostanza che impazzisca d'amore/odio per l'acqua.
E per concludere come il solito con qualcosa di pratico, ecco una vecchissima fotocellula che ho riesumato facendo un po' di scavi archeologici nello scatolone delle mie valvole elettroniche. |


|
Post n°342 pubblicato il 07 Febbraio 2016 da paoloalbert
Ancora un quadro? Ma non siamo in Chimica sperimentale?
Breton Woman in Prayer (1894) I cromati metallici insolubili sono stati usati fino ad alcuni decenni fa, fino a quando la sensibilità verso l'impiego di sostanze meno tossiche ha preso il sopravvento. I cromati di bario e stronzio sono abbastanza simili, ma con tonalità diverse: molto limone e molto freddo il primo, sempre chiaro ma più caldo il secondo. Ho mescolato, a pH neutro ed in quantità stechiometriche, una soluzione di cromato di potassio K2CrO4 ed una di nitrato di stronzio Sr(NO3)2, ottenendo il precipitato giallo di cromato di stronzio SrCrO4.
|


|
Post n°341 pubblicato il 02 Febbraio 2016 da paoloalbert
|
|
Post n°340 pubblicato il 16 Gennaio 2016 da paoloalbert
Tutti noi sappiamo che "LA CHIMICA" è vista da una generica signora Adele (e dai giornalisti del TG) più o meno come il fumo negli occhi e le bollette del gas. -C7H8, toluene. - C2H4O2, acido acetico. |
|
Post n°339 pubblicato il 06 Gennaio 2016 da paoloalbert
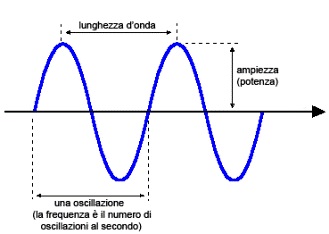
Una ventina di anni fa i televisori a schermo piatto erano da poco apparsi, poco diffusi e molto costosi, una specie di sogno futuristico più o meno come si vede nel film Farenheit 451 del 1966.
- di conseguenza, misurando la distanza di movimento della testa tra un minimo e l'altro si poteva risalire alla lunghezza d'onda, e da questa alla frequenza. |
|
Post n°338 pubblicato il 28 Dicembre 2015 da paoloalbert
|

|
Post n°337 pubblicato il 23 Dicembre 2015 da paoloalbert
|

|
Post n°336 pubblicato il 14 Dicembre 2015 da paoloalbert
A Monet piacevano le ninfee, sicuro.
E gli piaceva pure il color violetto, che troviamo sempre, naturalmente ora più ora meno, nel suo coinvolgente impressionismo che da solo vale il viaggio al museo, dove che sia.
I pigmenti violetti non sono tanti, anche se in ogni caso il viola lo si può ottenere con l'opportuna mescolanza del blù e del rosso; tuttavia la sostanza ottenuta da Salvetat certo portò un notevole contributo alle materie prime a disposizione dei pittori del periodo romantico e successivi. Mi sono messo per un attimo e immodestamente nei panni di monsieur Salvetat e ho provato a sporcare un po' di provette, naturalmente di corsa perchè d'inverno nel mio lab fa un freddo cane. 3 CoCl2 + 2 KH2PO4 + 4 KOH -> 2 Co3(PO4)2 + 6 KCl + 4 H2O In una prima fase ho salificato il KH2PO4 con KOH per ottenere il sale tripotassico controllando di non superare pH 12 (pH del sale dissociato) e poi ho aggiunto quest'ultimo alla soluzione di cloruro di cobalto, sempre tenendo conto delle rispettive quantità stechiometriche, che per semplicità questa volta ometto completamente. Ecco il mio risultato, immortalato in un fortunato momento di piacevole sole invernale:
E l'arseniato? Anche questo mi piacerebbe provar a fare (chissà se...).
|



|
Post n°335 pubblicato il 02 Dicembre 2015 da paoloalbert
"Similia similibus solvuntur!" Ma lasciamo perdere la frasetta latina di cui sopra e prendiamone un'altra, probabilmente ancora più celebre, quella inventata dal dottor Samuel Hahnemann a fine '700. Essendo un ferreo galileiano convinto, naturalmente non posso credere all'omeopatia fin che essa non sarà dimostrata scientificamente; sarebbe facilissimo applicare i consueti metodi di test per i farmaci, ma è chiaro che mai saranno applicati per i fin troppo ovvi motivi che cominciano con il simbolo del $ (da noi €) e per i risulltati, che sarebbero scontati. Ma voglio dire perchè IO non credo all'omeopatia. Significa semplicemente che in un granuletto c'è... c'è... stammi bene a sentire... ciò che resta dopo aver disperso mettiamo UN GRAMMO di Ignatia amara in MILLE MILIARDI di TONNELLATE di zucchero. -Che stai a dire, zotico ignorante- dice la gentile signora che mi legge [almeno una ci sarà, sono sicuro :-)] -che fa effetto non è la concentrazione ma la "dinamizzazione", la "memoria dell'acqua", quelle cose lì... qui non siamo in chimica! |
|
Post n°334 pubblicato il 16 Novembre 2015 da paoloalbert
Gino e Aldo si trovano per caso in Slovenia dalle parti di Kobarid (Caporetto) e chiacchierano del 1917.
Il sussurro del Kaiser all'entrata della miniera di Raibl semmai io lo interpreto molto liberamente come un affettuoso ringraziamento verso quei minatori che persero la vita (prima austroungarici e poi italiani, il sussurro vale per tutti!) estraendo il prezioso minerale di piombo e zinco dalle viscere della montagna. In che modo giocò il tiro? La miniera di Raibl è strutturata su molti livelli, il più profondo dei quali va a sfociare dal 1899 con una lunghissima galleria di scolo delle acque nell'attuale Slovenia, presso Log pod Mangartom (Bretto). Ebbene, da questa galleria il Generale Otto von Bulow (assieme a quell'altra VOLPE di Erwin Rommel) fece transitare in gran segreto nell'ottobre del '17 quasi 450mila soldati e 240mila tonnellate di tonnellate di materiali per sorprendere fulmineamente ed in maniera inaspettata l'esercito italiano sul fronte dell'Isonzo, determinandone in pochi giorni la disfatta e l'arretramento precipitoso fino al Piave. La cosa più curiosa di questa miniera è che a metà della famigerata galleria di Bretto dal 1947 fino al 1969 nelle viscere della terra passava uno dei confini di stato Italia-Iugoslavia e che un Carabiniere ed un Finanziere controllavano quei minatori che col microscopico trenino transitavano il VALICO DI FRONTIERA che dalla terra di Tito venivano a lavorare da noi. Nel 1990 la miniera chiuse definitivamente i battenti e oggi la si visita come turisti.
Purtroppo (anche questa volta!) il ferro la fa da padrone sotto forma della solita rugginosa limonite (ossido idrato di ferro FeO(OH).nH2O), mentre le tracce di galena (solfuro di piombo PbS) e blenda (solfuro di zinco ZnS) non si notano quasi per nulla nella fotografia, e devo dire che anche nel campione non sono certo abbondanti. |


Inviato da: paoloalbert
il 02/05/2021 alle 21:53
Inviato da: Bepi1249
il 15/04/2021 alle 14:11
Inviato da: Tanner85
il 06/02/2021 alle 00:01
Inviato da: poetryclub
il 04/10/2020 alle 23:00
Inviato da: poetryclub
il 04/10/2020 alle 20:58