CHIMICA sperimentale
Esperienze in home-lab: considerazioni di chimica sperimentale e altro
|
Post n°153 pubblicato il 20 Dicembre 2011 da paoloalbert
Vero che sono sempre complicate e aggrovigliate le associazioni di idee?
Che è successo? Improvvisa fusione neuronica nel mio brain? Intossicazione acutissima da composti cianosolforati? No, molto più semplice, adesso spiego il motivo di tale strana associazione mentale che di solito mi viene in questo periodo. Ecco, ora tutto si spiega, vero?
Mettere la senape nella mostarda non è semplice, come diceva la buon'anima di mia nonna: ce ne deve andare la giusta quantità, nè troppo, chè altrimenti il prodotto rischia di diventare una brace ardente che ti leva il fiato, nè troppo poco perchè ciò porta ad una massa fruttosa solo dolce e inconcludente. Nei prodotti commerciali, pur buoni, (quelli del nostro pacco lo sono) trovo difficile riscontrare questo giusto equilibrio tra frutta e senape e noto ogni anno con stupore che talvolta si tende ad esagerare in maniera sciocca con l'isotiocianato, come se si volesse virtualmente assassinare l'ignaro consumatore togliendogli il respiro. Il vasetto dovette rimanere aperto (ben protetto, ma aperto) fino a inverno inoltrato per poter essere consumato, sfruttando la nota volatilità dei tiocianati alchilici; inutile dire che il titolare dell'azienda produttrice, da me gentilmente invitato a casa mia a mangiarsi un pezzetto del suo mandarino candito, non si è mai presentato... sicuramente sapendo a cosa andava incontro. Si dirà che forse sono io che sono troppo delicato! |
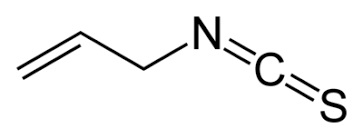
 Tutti sanno che nella vera mostarda, quella da intenditori, (quest'anno era quella mantovana) ci va la senape e la senape contiene appunto l'
Tutti sanno che nella vera mostarda, quella da intenditori, (quest'anno era quella mantovana) ci va la senape e la senape contiene appunto l'|
Post n°152 pubblicato il 16 Dicembre 2011 da paoloalbert
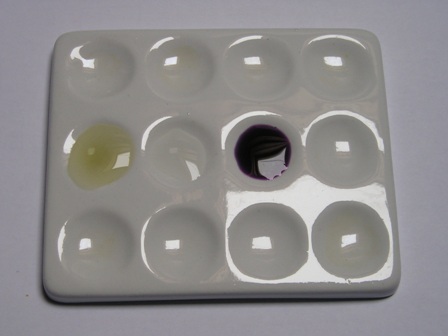
Ed eccoci finalmente al saggio completo del simpatico Sor Lasagna: dobbiamo trovare nel lab una sostanza che contenga tutti e tre insieme gli elementi "sensibili", cioè zolfo, azoto e alogeni, oltre naturalmente a carbonio, idrogeno e ossigeno tipici delle sostanze organiche.
La foto mostra le tre soluzioni che ho messo nei pozzetti per questa prova: a sinistra la prova in bianco con acqua e nitroprussiato, al centro la soluzione di Lassaigne da sola e a destra l'aggiunta alla medesima del nitroprussiato.
Le foto mostrano la soluzione prima e dopo l'aggiunta di AgNO3: mi pare che non ci siano dubbi nemmeno questa volta, anche il cloro c'è! |



|
Post n°151 pubblicato il 12 Dicembre 2011 da paoloalbert
Questa prima "lasagnata" (ved. post precedente) l'ho fatta come prova dimostrativa per la rilevazione dell'azoto, pertanto non sarà una vera ricerca ma una conferma, dato che sono partito volutamente da una sostanza ben ricca in azoto (circa il 47%!) per rendere sicuramente evidenti i risultati. La sostanza azotata e il sod io Il tubetto pronto per la pirolisi Le foto mostrano questi preparativi "statici", mentre lascio alla fantasia di chi legge immaginare le fasi "dinamiche", ovvero l'arroventamento del tubetto sul bunsen e la sua "decisa" reazione quando lo si butta in acqua. Andiamo a verificare.
Aggiunta di solfato ferroso Messi in una beutina circa 15 ml (ne basterebbero molti meno) della soluzione prima ottenuta, aggiungere una puntina di spatola di FeSO4 e mescolare; nell'ambiente così basico si forma subito un precipitato gelatinoso grigio verdastro di composti ferrosi, ma si forma anche il complesso ferrocianuro sodico Na4[Fe(CN)6] solubile e incolore, quindi non visibile.
Aggiungere una goccia di acqua ossigenata (o anche insufflare dell'aria per un po' di tempo) per ossidare parzialmente a ferrico il solfato ferroso, in modo da avere in soluzione anche ioni -Fe3+.
Formazione di Fe4[Fe(CN)6]3 Il residuo di Blù di Prussia Le foto mostrano la filtrazione e l'abbondantissimo residuo di ferrocianuro ferrico rimasto sulla carta da filtro, conferma assai evidente (lo sapevamo già!) che il campione in oggetto conteneva azoto e quindi aveva generato NaCN. |





|
Post n°150 pubblicato il 08 Dicembre 2011 da paoloalbert
In chimica organica analitica (classica) il saggio di Lassaigne è una pietra miliare: con una procedura semplicissima permette di determinare se una sostanza ignota contiene zolfo, azoto o alogeni, singolarmente o anche tutti insieme.
- il solfuro di sodio viene riconosciuto ponendo nel pozzetto di una piastrina in porcellana una decina di gocce di soluzione e aggiungendo, senza mescolare, qualche cristallino di sodio nitroprussiato Na3[Fe(NO)(CN)5]. - il cianuro di sodio va evidenziato (previa eliminazione preliminare dello zolfo se era presente) in questo modo: - gli alogenuri di sodio (-Cl, -Br, I) vengono ricercati (anche qui previa eliminazione dello zolfo) acidificando qualche ml di soluzione con acido nitrico non in eccesso e aggiungendo una decina di gocce di nitrato d'argento AgNO3 0,1 M. |


|
Post n°149 pubblicato il 06 Dicembre 2011 da paoloalbert
Non è vero che ho proprio distrutto totalmente la storica radio a galena di mio padre fatta negli anni di guerra: ne ho ancora i pezzi fondamentali e volendo la potrei addirittura ricostruire quasi come in origine.
Non mi addentro volutamente in nessun particolare tecnico, ne ho già discusso anche troppo nella scorsa presentazione.
Cosa si sente con questa radio? E' vero che i servizi radio (in OM e OC) sono stati fortemente ridimensionati per motivi economici da quasi tutti i principali paesi, ma tra ridimensionare un servizio pubblico e distruggerlo la differenza è abissale. Tutto sufficiente, nel 1944, per sentire ogni sera le famose quattro note della quinta sinfonia di Beethoven, che suonano esattamente come la lettera V telegrafica del Morse, DI-DI-DI-DAAA... la famosa V di Victory. |


 L'elemento chiave di questa radio è il
L'elemento chiave di questa radio è il  Dagli anni '50 in poi la galena è stata sostituita da un
Dagli anni '50 in poi la galena è stata sostituita da un |
Post n°148 pubblicato il 04 Dicembre 2011 da paoloalbert
|
|
Post n°147 pubblicato il 29 Novembre 2011 da paoloalbert
L'antefatto
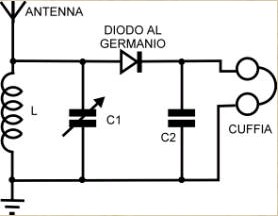
Dove si legge "diodo al germanio" si sostituisca il componente (che allora non esisteva) con l'originale "cristallo di galena" (PbS). Quella minimissima energia elettromagnetica che arriva... diciamo da Londra, entra nell'antenna (un filo lungo almeno una ventina di metri teso più in alto possibile), percorre la bobina L e si scarica a terra (un paletto conficcato in zona umida). Il circuito formato dalla bobina L e dal condensatore C in parallelo, diventa "risonante" su una determinata frequenza (quella della stazione che si intende ascoltare) quando il valori di L e di C sono opportunamente dimensionati, e per questo la capacità del condensatore è variabile con una manopola. In questo modo la componente in alta frequenza del segnale viene eliminata e rimane dopo il diodo solo la bassa frequenza audio derivata dalla modulazione del segnale (qui devo fare un piccolo ma doveroso omissis, altrimenti non ce la caviamo più!). Ma non ho ancora detto perchè si chiama radio a galena. Per tener fede allo spirito pratico-sperimentale del blog, concluderò queste riflessioni con qualche foto della "mia" radio a galena; purtroppo non è quella originale di mio padre, la quale, poverina, mai sarebbe potuta sopravvivere indenne ai massacranti "esperimenti" del giovane sottoscritto... |
|
Post n°146 pubblicato il 24 Novembre 2011 da paoloalbert
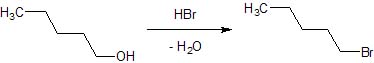
Allora oggi dobbiamo bromurare l'alcol amilico.
Materiali occorrenti
Porre a leggero riflusso per 2-3 ore, eventualmente con l'apparato per l'assorbimento acido come si vede in foto; ho notato però che praticamente quasi nulla sfugge dalla bocca del refrigerante. L'alchilbromuro si separa sopra la miscela acida e può essere separato facilmente.
Il bromuro di n-amile si presenta come un liquido limpido pesante (d. 1,22, p.e. 130°), di odore etereo pesante ma piacevole.
Ora il nostro alogenuro è pronto per la futura sintesi di un olezzantissimo tiolo! Quando si parla di tioli il problema "ambiente" si fa arduo, nel senso che è assolutamente improponibile fare queste preparazioni in laboratorio (se lo stesso non è munito di una efficientissima cappa!) perchè l'odore di questi composti col gruppo -SH (l'amilmercaptano è prorio uno di quelli giusti...) è davvero insopportabile ma soprattutto è molto persistente e dove si attacca rimane, vestiti compresi. |
 In pallone da 100 ml introdurre
In pallone da 100 ml introdurre 
 Predisporre quindi il refrigerante Liebig e distillare fin quasi a secchezza il grezzo preventivamente preparato, raccogliendo tra 127-132° (praticamente passa tutto in questo range).
Predisporre quindi il refrigerante Liebig e distillare fin quasi a secchezza il grezzo preventivamente preparato, raccogliendo tra 127-132° (praticamente passa tutto in questo range).
|
Post n°145 pubblicato il 21 Novembre 2011 da paoloalbert
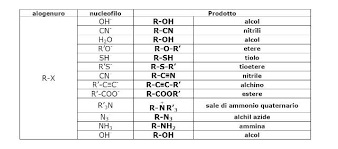
Gli alogenuri alchilici, di formula generale R-X (R = radicale alchilico, X = Cl, Br, I) sono dei reagenti fondamentali per la chimica organica; oltre agli importantissimi reattivi di Grignard possono dare una bella serie di sostituzioni nucleofile, che avvengono naturalmente ognuna nelle condizioni adatte: Nella seconda parte dirò della preparazione pratica del bromuro di n-amile. |
|
Post n°144 pubblicato il 18 Novembre 2011 da paoloalbert
Sembra strano, ma nella vecchia quanto ufficiosa nomenclatura chimica esistevano pure gli alberi, per lo più dedicati all'antica mitologia: e così c'è l'albero di Diana, l'albero di Marte, quello della Luna... e quello di Saturno.
In alto si vede parte dell'anodo e sotto il filo catodico ricoperto dall'alberello ad aghetti e squamette del metallo saturnino che sta crescendo.
In mancanza, ci metto questo elettroalberello. |


|
Post n°143 pubblicato il 14 Novembre 2011 da paoloalbert
Una volta (non l'anno scorso, un po' di più...) bastava andare dallo speziale, dire di essere perseguitati dai topi, chiedere una bustina di "arsenico" ed il gioco era fatto: ecco una decina di grammi di bianca anidride arseniosa a disposizione per qualsiasi evenienza! Ma poi, nel 1836, a dare una mazzata alle avvelenatrici (come si sa l'uso del veleno è da sempre prevalente appannaggio del gentil sesso!) è arrivato l'apparecchio di James Marsh...
Ecco dettagliatamente in cosa consiste e come veniva adoperato questo geniale congegno per la ricerca dell'arsenico, in uso dalla sua invenzione e fino ad una cinquantina di anni fa o anche meno. |

|
Post n°142 pubblicato il 11 Novembre 2011 da paoloalbert
Noi siamo ormai avvezzi per sovradosaggio mediatico ad ogni sorta di notizie di cronaca nera, tanto che non ci facciamo quasi più caso (se non nell'immediato), come drogati che devono aumentare sempre la dose di sostanza per risentirne gli effetti, i quali ben presto svaniscono. |
|
Post n°141 pubblicato il 08 Novembre 2011 da paoloalbert
Tutti sanno che il cromo prende il nome dal variegato colore dei suoi composti nei diversi stati di ossidazione.
La reazione che faremo avvenire è la seguente:
|

|
Post n°140 pubblicato il 03 Novembre 2011 da paoloalbert
Ho ritrovato il mio vecchio cannello ferruminatorio di quand'ero studente.
Prima di tutto, quando si rientra nella civiltà dopo lunghi viaggi in paesi remoti, occorre un bel bagno ristoratore; nel suo caso c'è voluto non un bagno ristoratore ma uno "restauratore", a base di... cartavetrata.
Bentornato fra i tuoi amici, cannello! |




|
Post n°139 pubblicato il 30 Ottobre 2011 da paoloalbert
No, purtroppo le femmine non c'entrano con il discorso che andrò a fare.
ecco come si presenta la valvola 6E5 e a fianco la vista della sommità illuminata, con ancora un settore d'ombra che si potrebbe restringere o allargare. Appropriato il nome "occhio", vero? Lo zoccolo della valvola è di tipo vecchio (anni '30) a 6 piedini, ma ne esiste anche una versione più moderna (anni '40) a otto piedini (in questo caso è detto "octal"). - pin 1: non collegato
|


|
Post n°138 pubblicato il 27 Ottobre 2011 da paoloalbert
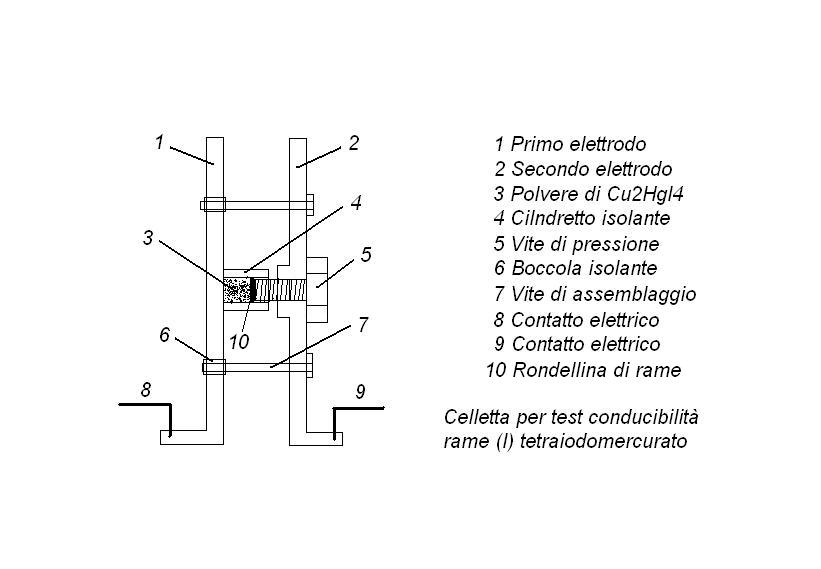
Nella discussione dedicata al termocromatismo del tetraiodomercurato rameoso Cu2HgI4 mi ero riproposto di allestire un setup adeguato alla misura delle variazioni della conducibilità elettrica relativa di questo interessante sale al variare della temperatura. Pertanto occorre pensare e assemblare un dispositivo che possa permettere una notevole compressione meccanica della sostanza in esame e contemporaneamente stabilire i relativi contatti elettrici senza incertezze.
Le immagini e il disegno in sezione spiegano la costruzione di questa celletta dedicata al Cu2HgI4.
Gli elettrodi sono ricavati da due profilati in alluminio a L, serrati assieme da quattro viti di tiraggio, isolate da una parte per mezzo di boccoline in nylon (quelle usate per l'isolamento dei transistor in TO3) in modo che le viti non costituiscano contatto elettrico tra le due piastrine.
Mi aspettavo uno scalino di transizione molto più netto intorno ai 70°, mentre ho notato che le variazioni sono macroscopiche ma progressive. Sottolineo fortemente che tutte queste misure sono RELATIVE, in funzione stretta delle condizioni di lavoro alle quali io ho operato, ma che comunque rendono bene l'idea che mi ero proposto, cioè di evidenziare "le variazioni" e non i valori assoluti, che in questo caso non sono di alcun interesse. Per curiosità ho testato anche il comportamento in media frequenza (fino a 100 KHz) di questa sostanza, senza alcun risultato significativo in semiconduttività: solo pura resistenza ohmica. Per la spiegazione del fenomeno rimando a quanto detto nel post precedente, trovando facilmente in rete ragione del fatto che ad un certo punto con l'aumento della temperatura gli ioni rame si mettono a saltellare di qua e di là nel reticolo cristallino in cerca di "buche", e così facendo (ricordo che --> cariche elettriche che si spostano = corrente!) rendono questa sostanza conduttiva con queste caratteristiche. |




|
Post n°137 pubblicato il 22 Ottobre 2011 da paoloalbert
Il Carnevale della chimica di fine ottobre, ospitato dal blog di Popinga, ci porta in tavola un argomento sfizioso: "La chimica in cucina", lasciando come il solito ai partecipanti una trattazione del tema molto discrezionale. Per non appesantire il discorso ometto i calcoli per risalire al numero di iodio del mio olio, che comunque mi ha dato un valore di 88, in linea con i valori dell'olio di oliva, che possono variare a seconda del tipo e della provenienza tra 80 e 88. (Nel mio caso si trattava di un olio della riviera gardesana, che detto per inciso è un'eccellenza...). |
|
Post n°136 pubblicato il 18 Ottobre 2011 da paoloalbert
Alcune sostanze si comportano in modo strano a seconda delle condizioni a cui sono sottoposte: c'è chi mostra fluorescenza, chi fosforescenza, chi piezoelettricità, chi piezoluminescenza, chi termoconduttività... ecc.... e c'è chi mostra il fenomeno del termocromatismo (o termocromismo), cioè il colore mostrato dalla sostanza è funzione della temperatura.
Le caratteristiche di questo interessante composto iodo-cupro-mercurico non finiscono qui: come dicevo all'inizio, esso al variare della temperatura mostra anche una notevole variazione di conduttività elettrica, che ho voluto investigare quantitativamente nei limiti dei miei mezzi, costruendo un piccolo dispositivo espressamente dedicato a questa prova.
|


|
Post n°135 pubblicato il 14 Ottobre 2011 da paoloalbert
Come dico spesso, ogni tanto arriva inevitabile la sintesi di un estere. Andiamo quindi ad apparecchiare la cucina: lo chef richiede oggi poca roba, è un piattino estivo molto semplice e fresco, di sicuro risultato e di buon gradimento... olfattivo. Allora, in barba a quei "chimici teorici" che non volendo sporcarsi le mani paragonano i chimici sperimentali a dei cuochi, mi dichiaro apposta cuoco chimico ruspante e così procedo:
-In un pallone da 250 ml porre 100 ml di acido acetico, 20 ml di metanolo e 4 ml di H2SO4 concentrato. Dati i vicini punti di ebollizione del metanolo (64,7°) e del metilacetato (57°) ho usato un grande eccesso di acido rispetto all'alcool (più di 3:1) in modo da spostare l'equilibrio a destra il più possibile verso una esterificazione abbastanza completa ed aver meno problemi nella separazione finale. Stavolta non ho messo fotografie della sintesi, perchè si assomigliano tutte e hanno poco significato; in ogni caso... la saga degli esteri continua! |
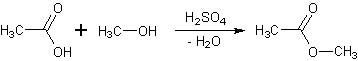
|
Post n°134 pubblicato il 11 Ottobre 2011 da paoloalbert
In occasione del post sui ricordi di scuola ho accennato all'impiego del cannello ferruminatorio; naturalmente si parla a proposito di quegli studenti di chimica che, come me, frequentavano qualche anno fa laboratori dove ancora nessun "apparecchio con la spina" aveva preso piede. I piaccametri li ricordo benissimo perchè, per me che ero stralunato anche nei riguardi dell'elettronica, avevano come indicatore la valvola 6E5GT, uno di quegli "occhi magici" delle antiche radio, con un iride luminescente verde che si apriva e chiudeva secondo l'intensità del segnale, nel caso specifico secondo le indicazioni della sonda al calomelano. L'amico Simone mi ha chiesto come si usavano: ecco come. Le riduzioni sul carbone al cannello ferruminatorio facevano parte dei cosiddetti "saggi preliminari" di una analisi inorganica. Ognuno di noi aveva a disposizione (comprandolo, è chiaro!) un bel blocchetto di carbone di legno di tiglio, una specie di mattoncino resistente lungo un palmo. Il cannello ferruminatorio non era altro che un tubicino di ottone lungo una trentina di centimetri, rastremato in più sezioni e con la punta piegata a L; soffiando nella parte di maggior diametro, si poteva dirigere il dardo concentrato dove si voleva. Essendo il carbone poroso, le sostanze facilmente fusibili come gli alcali sono assorbite, e le altre trasformate prima in carbonati, poi in ossidi e per ulteriore riduzione in metallo. Metalli volatili come Zn, Cd, As, si ossidano comunque e danno un'aureola caratteristica nella direzione opposta a cui si soffia: gialla a caldo e bianca a freddo per lo zinco, marrone per il cadmio, bianca e volatile per l'arsenico (oltre all'odore agliaceo). Si possono riconoscere al carbone anche alcuni sali, per esempio i nitriti, nitrati, clorati, che riscaldati producono microscopiche "deflagrazioni" e certi altri (NaCl per es.) che producono "crepitazione". Naturalmente dopo un po' di tempo il mattoncino di tiglio era tutto bruciacchiato e pieno di aloni e residui di ogni tipo e andava cambiato... ma nel frattempo l'anno scolastico volgeva al terzo trimestre e si passava magari a qualcosa di più "tecnologico", magari andando nel divertentissimo (almeno per me!) Laboratorio di Chimica Organica, al quale ancora oggi assegno con onore le iniziali maiuscole. Tutto molto empirico vero? Certo, molto empirico se vogliamo, ma anche molto divertente!
|
Inviato da: paoloalbert
il 02/05/2021 alle 21:53
Inviato da: Bepi1249
il 15/04/2021 alle 14:11
Inviato da: Tanner85
il 06/02/2021 alle 00:01
Inviato da: poetryclub
il 04/10/2020 alle 23:00
Inviato da: poetryclub
il 04/10/2020 alle 20:58