CHIMICA sperimentale
Esperienze in home-lab: considerazioni di chimica sperimentale e altro
|
Post n°113 pubblicato il 24 Giugno 2011 da paoloalbert
A proposito della crisoina dell'altra volta, e parafrasando un noto presentatore, si potrebbe dire che "sorge spontanea una domanda": perchè questa sostanza è un colorante e mille altre non lo sono?
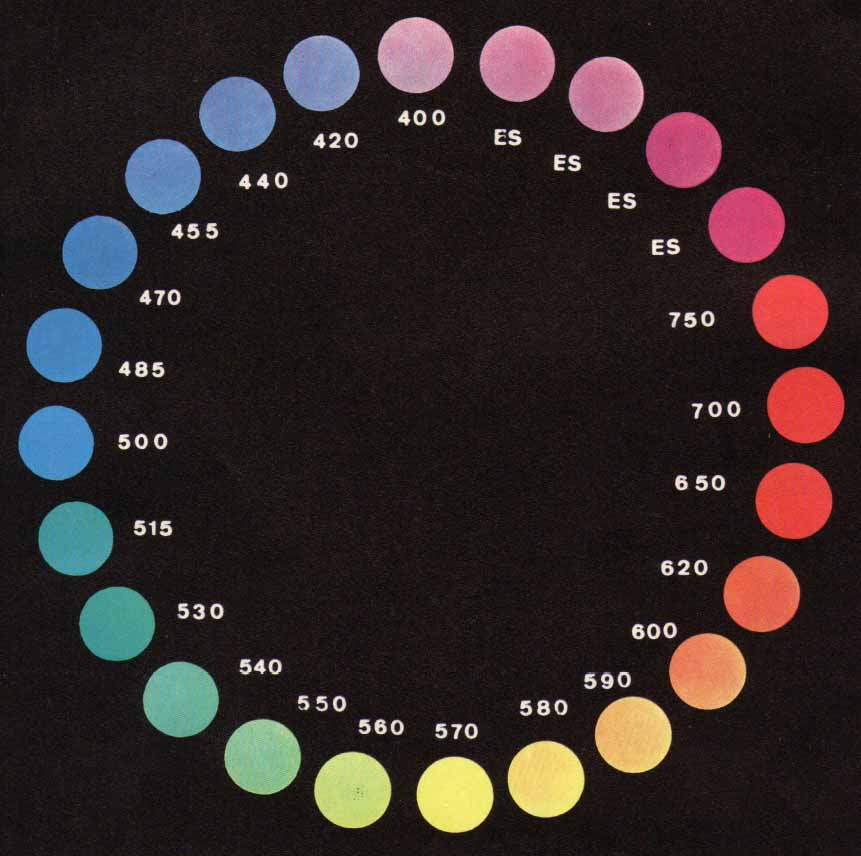
Siccome l'occhio umano non è in grado di distinguere i vari colori quando viene colpito contemporaneamente da radiazioni di λ diversa, oltre ai colori "puri", esistono infinite combinazioni diverse, che danno la sensazione della tonalità cromatica a seconda dell'assorbimento più o meno selettivo da parte della sostanza colorata. La presenza di una particolare struttura e di certi raggruppamenti atomici nella molecola di un composto chimico conferisce la capacità di assorbire selettivamente luce visibile e rendere quel composto colorato. Succede che se in una molecola sono presenti doppi legami coniugati (-C=C-C=C-) gli elettroni risultano maggiormente delocalizzati, con conseguente ulteriore diminuzione energetica tra un livello e l'altro e più facile eccitabilità da parte di una radiazione visibile. La vecchia teoria classica di Witt (più intuitiva in un semplice contesto come questo) affema che una molecola per apparire colorata deve possedere almeno un gruppo "cromoforo" e percolorare almeno un gruppo "auxocromo". :C=C: etilenico Un auxocromo permette sia la fissazione del colorante ad un substrato sia di aumentare la λ della radiazione assorbita rendendo colorata una molecola che senza di esso assorbirebbe nell'UV, oppure di spostare verso il rosso la tonalità (diverso effetto di una stessa causa). Per concludere queste semplici riflessioni, torniamo alla crisoina e verifichiamo: -c'è il fondamentale gruppo -azo? Sì! Jawohl! esclama allora Herr Otto Witt,... perchè mai la crisoina non avrebbe il sacrosanto diritto di farti una bella macchia gialla sulla camicia?
|
|
Post n°112 pubblicato il 16 Giugno 2011 da paoloalbert
Dopo qualche divagazione "elettrostatica" (ma fatta con molta soddisfazione!) torniamo a noi, ovvero alla chimica sperimentale.
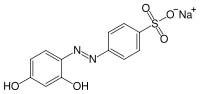
- Acido solfanilico p-NH2-C6H4-SO2-OH - In un becker da 100 ml sciogliere 2,5 g di acido solfanilico e 1 g di NaOH in 40 ml di acqua.
Lavare con soluzione satura di sale, aspirando bene per eliminare quanto più liquido possibile e poi essicare lentamente e con fatica all'aria.
La crisoina secca si presenta come una polvere color ruggine, discretamente solubile in acqua e in etanolo; tinge i tessuti in colore dai toni caldi dal giallo all'ocra ed è anche un indicatore di pH nel range basico 11-12,7 (giallo per pH <11 e rosso per pH >12,7
|
 Il problema maggiore a questo punto è che la separazione, perchè la soluzione alcalina di crisoina e la
Il problema maggiore a questo punto è che la separazione, perchè la soluzione alcalina di crisoina e la  La maggior parte della
La maggior parte della 


|
Post n°111 pubblicato il 11 Giugno 2011 da paoloalbert
Ecco la macchina di Van de Graaff completa!
(Una doverosa conclusione: da tempo il mio amico Guglielmo mi incoraggiava a fare questa macchinetta; siccome ora sta passando un periodo personale veramente difficile, a lui dedico questo lavoro). |

|
Post n°110 pubblicato il 08 Giugno 2011 da paoloalbert
Se è vero che alla fine tutti i nodi vengono al pettine, è arrivato finalmente al pettine anche questo nodo: la costruzione di una macchina Van de Graaff, che avevo in progetto da tempo immemorabile.
Motore e rullo inferiore Trasformatore per il motore
Cinghia e rullo superiore Il pettine interno alla sfera
Premetto subito che (a meno di non fare un modellino scalcagnato) non è una costruzione facile come forse potrebbe sembrare: una buona attrezzatura meccanica e precisione nell'esecuzione sono indispensabili. Come materiale per tutta la struttura, a parte la base di sostegno, ho usato il plexiglass, che è un materiale esteticamente bello e adatto per lavori in alta tensione ma è schifosamente scomodo da lavorare; tuttavia avevo deciso di usare questo e con questo ho proseguito. diametro in mm = tensione in kilovolt Il pettine inferiore per l'effetto corona è realizzato sagomando a punte un lamierino di rame; è singolare il fatto che il massimo rendimento si ottiene posizionandolo non in corrispondenza del rullo ma spostandolo una decina di cm più in alto, lungo il nastro. |




|
Post n°109 pubblicato il 04 Giugno 2011 da paoloalbert
Robert Jemison Van de Graaff nacque a Tuscaloosa, Alabama, il 20 dicembre 1901.
Nel 1935 V.d.G. ottenne il brevetto per la sua invenzione, destinata, oltre che come acceleratore di particelle, per la produzione di raggi X penetranti per la cura di tumori. "un dispositivo che ha permesso un immenso progresso nella ricerca nucleare". |

|
Post n°108 pubblicato il 01 Giugno 2011 da paoloalbert
Accennavo l'altra volta all'idiosincrasia mediatica verso la chimica e tutto il suo mondo, indotta da un'informazione e da una cultura scientifica media che non ardisco definire. State a sentire perchè la ballata è bella ed i protagonisti sono tanti e molto articolati... nelle loro ramificazioni laterali, se così vogliamo dire. Allora, nella bomboletta c'è dentro (maestro, vai con la musica):
Troppi perchè che non avranno mai una risposta. |
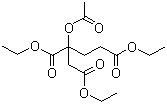
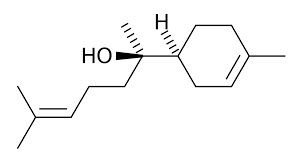
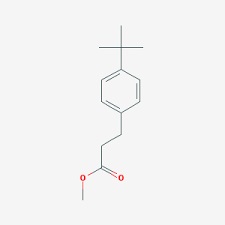
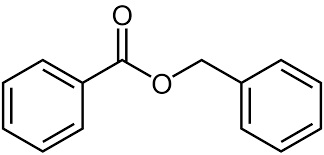
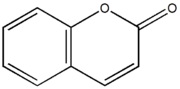
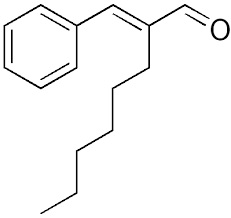
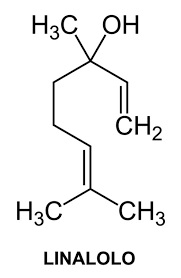

|
Post n°107 pubblicato il 28 Maggio 2011 da paoloalbert
Mi è stato chiesto da un lettore di questo blog dove trovo un facile reagente, l'acido fosforico. |
|
Post n°106 pubblicato il 27 Maggio 2011 da paoloalbert
Nel blog dell'amico Marco Capponi si è parlato recentemente di quella storica miniera della Val Imperina nell'Agordino, dalla quale, partendo da una pirite debolmente cuprifera, veniva estratto il rame che fin dalla notte dei tempi suppliva al fabbisogno di questo fondamentale metallo la Serenissima Repubblica di Venezia.
|

|
Post n°105 pubblicato il 14 Maggio 2011 da paoloalbert
Stavolta solo chimica applicata, poche parole di contorno e tante di procedura.
Il grumo molto lentamente si disgrega e si scioglie, lasciando alla fine solo un liquido biancastro, che va filtrato; un po' di selenio si perde anche in questa fase, ma è sempre meglio portarsi dietro meno porcherie possibili.
Diluire il filtrato con altrettanta acqua e poi aggiungere lentamente ammoniaca, sempre mescolando energicamente; ad ogni aggiunta precipita l'idrossido di cadmio bianco, che mescolando si ridiscioglie finchè l'ambiente è sufficientemente acido; quando non si sciolie più, tornare leggermente indietro con qualche goccia di HCl, fino a riottenere una soluzione limpida e e senza più HNO3 libero.
Porre la soluzione in un recipiente alto e sottile (un cilindro graduato) per favorire il massimo contatto con il gas e far gorgogliare una sufficiente quantità di SO2, facendola sviluppare in un pallone dalla reazione tra metabisolfito K2S2O5 e HCl; scaldando leggermente il pallone di reazione si ottiene una buona corrente di anidride solforosa, portata al fondo del cilindro da un tubo di vetro.
Continuare con la SO2 se si vuole ridurre con questo metodo, oppure interrompere e provare con la procedura all'idrazina, molto più comoda perchè avviene in fase omogenea.
Alla soluzione residua aggiungere mescolando 4 g di idrazina solfato e scaldare appena appena se si vuole ottenere prevalentemente selenio rosso, lasciando in riposo una giornata.
Per ebollizione l'allotropo rosso si trasforma e si aggrega nella forma grigia, molto più facilmente separabile. In ogni caso filtrare, lavare accuratamente e lasciar asciugare.
Per fusione in capsula di porcellana si ottiene il selenio nero;
per raffreddamento molto lento il selenio si trasforma nell'ulteriore allotropo metallico (selenio grigio, non fatto). |








|
Post n°104 pubblicato il 10 Maggio 2011 da paoloalbert
Il titolo è molto provocatorio... è ovvio che il selenio al massimo si estrae, mica lo si "fa", ma anche l'estrazione hobbistica di questo elemento non càpita tutti i giorni.
Il rosso di cadmio è reperibile in qualche colorificio professionale che tratta pigmenti originali in polvere, come si usava una volta quando gli artisti (o altri operatori del settore) si facevano da soli i colori miscelando bellissime polverine colorate assieme ad olio di lino o simili disperdenti. ---oooOOOooo--- Allora, pesare 10 g di rosso di cadmio, metterli in una beuta da 150 ml su agitatore magnetico e aggiungere... |

|
Post n°103 pubblicato il 04 Maggio 2011 da paoloalbert
Perchè un post dedicato espressamente al selenio?
Gli appassionati di fotografia meno giovani (chiamiamoli così...) ricorderanno il balletto che si doveva fare prima di ogni scatto tra la macchina fotografica e quell'aggeggio chiamato esposimetro, che forniva, grazie al selenio, la giusta esposizione su cui regolare manualmente tempi e diaframmi...
Ora, di questi tempi, mi son messo a giocare addirittura col selenio come 34° elemento della Tavola Periodica: la prossima volta lo farò in concreto. |
 Il selenio metallico al buio conduce poco la corrente elettrica, ma se illuminato la sua conducibilità aumenta di un migliaio di volte per effetto fotoelettrico; tale effetto è stato sfruttato fin nelle prime
Il selenio metallico al buio conduce poco la corrente elettrica, ma se illuminato la sua conducibilità aumenta di un migliaio di volte per effetto fotoelettrico; tale effetto è stato sfruttato fin nelle prime 
 Col selenio (o meglio con i
Col selenio (o meglio con i |
Post n°102 pubblicato il 28 Aprile 2011 da paoloalbert
Leggendo tra le righe con spirito critico i risvolti esclusivamente tecnici del lavoro descritto nel post precedente, rilevo un particolare poco chiaro nelle righe dell'Ambrosioni; forse si sarà trattato di un'omissione o di una svista dell'Autore, oppure di qualcosa che mi sfugge. |
|
Post n°101 pubblicato il 26 Aprile 2011 da paoloalbert
Dopo le quattro puntate precedenti, non val la pena di rilassarsi un pochino leggendo un paio di paginette dell'Ambrosioni, come quando si va in bagno e si cerca qualche lettura di poco impegno?
-L'azzurro di Berlino conosciuto in commercio altresì coi nomi di bleu, ossia azzurro Prusso, e dai Chimici Idrocianato di tritossido di ferro, trae il suo nome dalla città, ove per la prima volta è stato inventato, e fabbricato. |

|
Post n°100 pubblicato il 23 Aprile 2011 da paoloalbert
I don't know how to love him... -Non so come amarlo- cantava la struggente Ivonne Elliman nel 1970 in uno dei più significativi momenti dell'opera pop Jesus Christ Superstar.
|
|
Post n°99 pubblicato il 19 Aprile 2011 da paoloalbert
Dopo essere stato preventivamente testato al banco mentre cresceva (ved. foto nel post precedente), ho slegato dal lettino il circuito "piccolo Frankestein" liberandolo dalla miriade di fili che lo alimentavano e l'ho piazzato in una scatoletta che avevo, in modo che fosse indipendente e potesse cominciare a muoversi autonomamente facendo il lavoro che doveva fare.
Le foto, fatte in qualche modo di sera, rendono un'idea del marchingegno finito durante una misura sotto descritta.
Prove di assorbimento |


|
Post n°98 pubblicato il 17 Aprile 2011 da paoloalbert
Dopo il lavoro di precisione abbastanza noioso ma indispensabile per la celletta, mi sono preso un po' più di soddisfazione con la progettazione e realizzazione del circuito. Sottolineo che occorre amplificare non il segnale, ma le "variazioni" del segnale stesso a seconda della concentrazione, e queste variazioni possono essere debolissime, anche se il segnale in assoluto è forte.
Nella foto si vede l'accrocco mentre è ancora in fase di progettazione, i componenti disassemblati belli distesi e pieni di fili come Frankestein, che dà i primi segni di vita leggendo l'assorbimento di una soluzione di sali di rame.
Le prime prove all'oscilloscopio mostrano che l'apparecchio come principio funziona e sono quelle che mi danno la forza di continuare nella sperimentazione! |



|
Post n°97 pubblicato il 14 Aprile 2011 da paoloalbert
Come dicevo nell'introduzione, tempo fa mi è venuta voglia di provare a fare un piccolo "spettroscopio/colorimetro" sperimentale, naturalmente senza alcun intendimento di costruire uno strumento di misura, ma con lo scopo di fare un "giocattolo", diciamo così, didattico che verificasse l'assorbanza relativa di uno ione (o di una molecola colorata) e anche per avere la scusa di sposare bene assieme i miei due hobbies principali (chimica ed elettronica).
La cuvetta è quel piccolo contenitore parallelepipedo di cui è noto il cammino ottico ed il materiale è di buona trasparenza; ci sono cuvette in vetro, a facce perfettamente parallele e calibrate (molto costose), e cuvette economiche in plastica: indovinare quali ho usato io... Le due protuberanze laterali in rame che si vedono in foto contengono uno l'elemento illuminatore e l'altro il ricevitore.
|

|
Post n°96 pubblicato il 10 Aprile 2011 da paoloalbert
Cercando in rete l'argomento "spettroscopia UV-visibile" si trova tutto quello che io ometterò di dire in questa sede, la quale, come ormai ben si sa, è dedicata quasi categoricamente alla realizzazione pratica di quello che la teoria propone. Solo due parole minime introduttive al mio esperimento (anche hardware!) che seguirà: la spettroscopia è una disciplina che riguarda quelle tecniche con le quali è possibile ottenere informazioni sulle proprietà strutturali dei corpi studiando l’interazione della materia con l’energia elettromagnetica, cioè la luce, visibile o meno.
Dopo questa doverosa premessa ULTRA-semplificata, ho provato a costruire un aggeggio che facesse vagamente quanto sopra: |
|
Post n°95 pubblicato il 05 Aprile 2011 da paoloalbert
Nel blog ci vuole ogni tanto uno stacchetto, magari d'arte (una musica, un'immagine...).
|


|
Post n°94 pubblicato il 03 Aprile 2011 da paoloalbert
Ironia della sorte, una volta abitavo vicino ad una località che si chiamava proprio "Silani"... una specie di microscopica Silicon Valley ante litteram: ma quelle quattro case non avevano a che fare con i composti del silicio! |
Inviato da: paoloalbert
il 02/05/2021 alle 21:53
Inviato da: Bepi1249
il 15/04/2021 alle 14:11
Inviato da: Tanner85
il 06/02/2021 alle 00:01
Inviato da: poetryclub
il 04/10/2020 alle 23:00
Inviato da: poetryclub
il 04/10/2020 alle 20:58