CHIMICA sperimentale
Esperienze in home-lab: considerazioni di chimica sperimentale e altro
|
Post n°93 pubblicato il 31 Marzo 2011 da paoloalbert
Lo scopo di questo esperimento lo si vedrà appieno nel prossimo post (anche se qualcuno lo potrà facilmente intuire). La reazione fondamentale da eseguire è la seguente: 4 Mg + SiO2 --> 2 MgO + Mg2Si in cui il magnesio si ossida a spese dell'ossigeno della silice ed il silicio si riduce da ossido ad elemento e si combina con l'eccesso di magnesio per dare il siliciuro Mg2Si. Preparazione della silice Come fonte di SiO2 ho usato sabbia comune, a dire il vero molto impura per calcio e ferro; non avendo a disposizione silice pura, ho lavato prima la sabbia con acqua e poi con HCl al 15% a caldo; in questo modo viene eliminato il calcare ed un po' di ferro. La procedura che segue è delicata e va eseguita con le dovute precauzioni e in ambiente adatto. Modifiche maggiorative nelle dosi indicate o umidità nei reagenti possono portare a reazione incontrollata o comunque pericolosa. Materiale occorrente: - silice SiO2 pura (in mancanza, prepararla come sopra) - mescolare 2 g di polvere di magnesio e 1,5 g della silice prima preparata (questa è in leggero eccesso per compensare la purezza) e porli sul fondo di una provetta asciuttissima e "sacrificale". Il motivo di questo termine lo si capirà alla fine dell'esperimento. Naturalmente quando si parla di combustione avvengono sempre altre reazioni secondarie indesiderate, che portano il siliciuro di magnesio ottenuto ad essere molto impuro, tuttavia sufficiente per gli usi di curiosità che ne andremo a fare.
Per ora mettiamo da parte il nostro nero siliciuro in attesa di vedere cosa sarà capace di fare la prossima volta. |

|
Post n°92 pubblicato il 27 Marzo 2011 da paoloalbert
Poco meno di un quarto di secolo fa (mannaggia quanto tempo...!), mi ero procurato un contatore Geiger ex militare dell'Esercito tedesco, sull'onda emozionale dell'evento di ЧернобЫл, anche se troppo tardi per poterlo provare in diretta sul fall out italiano di quel disastro nucleare. In questi tempi molto sfortunati per il Sol Levante, ho riesumato il mio vecchio articolo e rispolverato l'apparecchio, purtroppo tornato forzatamente di moda con il disastro di Fukùshima. Senza entrare nel funzionamento di un Geiger (ciò si trova facilmente altrove), dico solo due parole sullo schema: tutto ruota attorno al tubo sensibile alle radiazioni β + γ (FHZ76V); un transistor funziona come oscillatore per la generazione dell'alta tensione necessaria al tubo (500 V) e altri tre transistors costituiscono gli stadi di amplificazione per il pilotaggio del milliamperometro e dell'uscita in cuffia. I fondo scala di lettura dell'apparecchio sono rispettivamente 1 R/h, 25 mR/h, 0,5 mR/h, 10.000 impulsi/min, 320 impulsi/min e quindi l'unità di misura è espressa in mR/H (milliRoentgen/ora) ed in impulsi/min, che sono i classici ticchettii che si sentono nei film catastrofici quando viene inquadrato un Geiger. Per vedere e sentire lo strumento in funzione ho avvicinato per l'occasione al tubo sensibile un milliamperometro di un apparecchio aeronautico militare della seconda G.M., che ha i riferimenti che un tempo erano fosforescenti per poter essere letti al buio.
Sarei curioso, solo virtualmente s'intende!, di vedere dove sarebbe andata la lancetta dell'FH-40 se mi fossi trovato nel medesimo momento in qualche posto del nord del Giappone... |
 La radiattività letta con questo milliamperometro ex luminescente a contatto del tubo raggiunge picchi di
La radiattività letta con questo milliamperometro ex luminescente a contatto del tubo raggiunge picchi di 
|
Post n°91 pubblicato il 23 Marzo 2011 da paoloalbert
Mi trovo a possedere un po' di tornasole, quello classico, vegetale.
L'ho chiamato indicatore fuori moda perchè ormai è un ausilio chimico assolutamente obsoleto, sostituito produttivamente da molti altri indicatori molto più selettivi nel range di pH da controllare, e soprattutto dalle comodissime "cartine universali": sono dei lunghi rotolini di carta assorbente imbevuta di una speciale miscela di indicatori che cambiano una varietà di colori in un range molto ampio, tant'è che dalla tonalità di colore assunta si può apprezzare il valore del pH di una soluzione a step di una unità, da 1 a 12 e in un campo più ristretto anche con maggior definizione. Il tornasole (come quasi tutti gli indicatori singoli) ha due tonalità di colore: è rosso sotto pH 4,5 e viola-bluastro sopra pH 8,3; tra i due valori, cioè circa alla neutralità, è un brutto rosa smorto. Non sono riuscito a trovare una giustificazione certa dell'etimologia del nome italiano tornasole e francese (tournesol); il prefisso "torna" esprime chiaramente il senso di "voltare, girare" come i girasoli si volgono verso il sole, come del resto ricorda anche il prof. Guilizzoni nella sua "Etimologia di alcuni termini scientifico-tecnici".
|

 Questa sostanza naturale è un colorante ricavato da molti vegetali diversi della famiglia dei licheni, a seconda della zona geografica; principalmente si ricavava dalla
Questa sostanza naturale è un colorante ricavato da molti vegetali diversi della famiglia dei licheni, a seconda della zona geografica; principalmente si ricavava dalla 
|
Post n°90 pubblicato il 19 Marzo 2011 da paoloalbert
Parlando recentemente dell'acqua borica ho voluto accennare alla buon'anima di mia nonna Beatrice, la quale, grazie alla sua immancabile bottiglia di H3BO3 nell'armadio, ritengo che abbia in qualche modo contribuito all'impianto del virus chimico nell'allora verde e incontaminato DNA del sottoscritto.
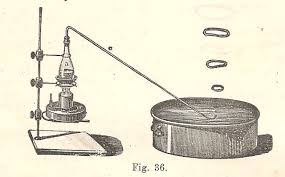
Il prof. S.Squinabol, illustre geologo di Rovereto, era certo una mente eclettica (una parte di merito va certo riconosciuta anche al prof. Cresci) perchè questo bel tomo targato 1898 contiene argomenti di chimica, di fisica, di mineralogia e di elettrologia, tutti descritti in maniera semplice e accattivante, e nello stesso tempo esauriente quanto basta agli scopi del libro, che era destinato agli alunni (nel caso dei miei bisnonni, di Rovereto) delle allora "scuole normali", corrispondenti alle classi superiori di oggi. Le immagini di questi libri sono tutti piccoli capolavori tecnici di disegno e ombreggiatura, che rendono l'oggetto rappresentato molto più significativo di una cruda fotografia; ne propongo due come esempio: la simulazione in laboratorio delle camere a piombo per la produzione dell'acido solforico
e la generazione artificiale dei "fuochi fatui" (ovvero delle fosfine PH3 e P2H4), con gli immancabili anelli di fumo che si innalzano dal recipiente pieno d'acqua!
Ma il libro è pieno di immagini simili, su ogni argomento trattato: dalla pistola di Volta innescata con una macchina elettrostatica, al fonografo di Edison fresco di invenzione, alla macchina di Carrè per fabbricare il ghiaccio, fino alla più spinta avanguardia: il telegrafo senza fili, fresco... di concezione! Potevo, io bimbo non ancora vaccinato a niente (ma certamente "presensibilizzato" per cause ignote), rimanere insensibile a reiterate e reiterate letture del libro del bisnonno? |


|
Post n°89 pubblicato il 16 Marzo 2011 da paoloalbert
Siamo in periodo di Carnevale (chimico, s'intende...): perchè non far sfilare virtualmente delle mascherine in costume d'epoca, travestite da Acque storiche? Faccio marciare queste "Acque storiche" così come me le sono ricordate, più o meno secondo l'uso che di questa "acqua" si faceva; non è detto che siano tutte formate letteralmente da H2O, (anzi nessuna lo è del tutto) e nemmeno che siano completamente obsolete: qualcuna di esse si usa ancora ed ha conservato il nome originale. Immaginiamocele vestite come meglio ci pare: taca banda!
Le soluzioni acquose degli alogeni sono abbastanza importanti per la chimica analitica: Acqua di cloro: è una soluzione di due volumi di cloro Cl2 in un volume di acqua (il cloro è poco in peso!); si forma un equilibrio ionico di Cl-, H+ e ClO-. Ha le stesse proprietà ossidanti del cloro gassoso, ma in forma blanda e maneggevole. Acqua di bromo: bella soluzione di colore rosso aranciato al 3% di Br2; usata soprattutto in chimica organica per il riconoscimento dei doppi legami, ai quali il bromo si lega con conseguente decolorazione del reattivo. Esisteva anche una sfuggente Acqua di iodio, utile per la conferma analitica dei bromuri col metodo all'argento.
Chiamo in questo modo le soluzioni acquose degli idrossidi dei metalli alcalino-terrosi. Acqua di calce: soluzione allo 0,5% di idrossido di calcio, Ca(OH)2; serve/serviva come liquido alcalino in varie occasioni, in arte, nel restauro, per regolare il pH degli acquari, ecc. Acqua di barite: soluzione al 5% di idrossido dai bario, Ba(OH)2; può essere ancora utile per la rivelazione o la conferma della presenza di anidride carbonica in un gas: il Ba(OH)2 in presenza di anidride carbonica (CO2) si trasforma in carbonato di bario BaCO3 insolubile e ben visibile. Per completezza c'era una volta anche la più esotica e suggestiva Acqua di stronziana, Sr(OH)2, di usi analoghi, dove il metallo alcalino-terroso è il simpatico Stronzio.
Le acque ad uso di farmacopea sono infinite se ci si vuol riferire anche ai decotti e soluzioni di sostanze curative di origine per lo più vegetale; in questa sede voglio ricordare invece solo un paio di acque prettamente chimiche, la cui presenza nelle domestiche "farmacie" risale fino a non molti decenni fa. Acqua borica: soluzione al 3% di acido borico, H3BO3, estratto soprattutto da alcune sorgenti toscane di vapore caldo (soffioni boraciferi); l’acqua borica veniva generalmente utilizzata per disinfettare leggere lesioni come eczemi e ustioni, e per bagni oculari. Ricordo che mia nonna ne aveva sempre una bottiglia pronta nell'armadietto... assieme a tante altre belle cose dei tempi che furono. Sicuramente devo qualcosa anche a mia nonna riguardo il virus chimico che mi ha contagiato sin da piccolo! Acqua vegeto-minerale: era una soluzione al 1% di acetato basico di piombo, Pb(CH3-COO)2.Pb(OH)2, ed era detta anche Acqua di Saturno; serviva per lenire i dolori dovuti a contusioni o slogature. Oggi questo zuccherino sale di piombo, anche allora sorvegliato speciale, non sarebbe certo lasciato libero...). Acqua epatica (anche Acqua zolfa): è una soluzione naturale di acido solfidrico H2S. Acqua tofana: mi prendo la libertà di aggiungere qui, anche se certamente medicinale non è, la cosiddetta Acqua tofana o Acquetta di Perugia: nonostante il nome carino e insospettabile, era una micidiale soluzione, in gran voga soprattutto nel periodo rinascimentale, di anidride arseniosa As2O3, con aggiunta di altre sostanze estremamente tossiche (sali di piombo, alcaloidi come cantaridina, atropina, aconitina) usata espressamente per avvelenare il prossimo. E' uno dei simboli degli antichi veleni, un tempo assai più usati di oggi per liberarsi degli scomodi concorrenti agli effimeri troni o, più modestamente, dei concorrenti... di letto!
Sono soluzioni di ipocloriti alcalini, utili prodotti per l'industria e per l'impiego domestico. Acqua di Javel: è una soluzione al 5% di ipoclorito di sodio, NaClO, ed è ancora ai giorni nostri sulla cresta dell'onda. Se potessimo andare in un ipotetico supermercato di un secolo fa, la normale candeggina si troverebbe sullo scaffale in una bella bottiglia di vetro con una arzigogolata etichetta belle epoque: "Acqua di Javel"! (dal nome del quartiere parigino Javel, nel quale lavorava a fine '700 il famoso chimico C.L.Berthollet). Acqua di Labarraque: idem come sopra, ma si dà un po' le arie da nobile! Anzichè essere plebeo ipoclorito di sodio era aristocratico ipoclorito di potassio KClO. Acqua ossigenata: il perossido di idrogeno H2O2 non è certo caduto in disuso e rimane un importantissimo prodotto industriale e reagente chimico. Come uso detergente/sbiancante l'acqua ossigenata viene oggi usata spesso sotto forma di perborato di sodio (NaBO2.H2O2.3H2O) , ormai anche lui sostituito dal percarbonato di sodio (Na2CO3.1,5H2O2), sostanze che in acqua calda liberano H2O2 e indirettamente ossigeno attivo, dalle proprietà disinfettanti e sbiancanti per il bucato.
Non mi riferisco alle deboli e dannose piogge acide, ma a sostanze che acide lo sono veramente in massimo grado. Acqua regia: L'acqua regia è una miscela composta da un volume di acido nitrico HNO3 e tre volumi di acido cloridrico HCl concentrati. I due acidi mescolati danno origine al cloruro di nitrosile NOCl, sostanza estremamente aggressiva nei confronti di quasi tutti i metalli; il suo nome deriva dalla sua capacità di sciogliere i metalli nobili, oro compreso, considerato dagli alchimisti il "re dei metalli" in quanto praticamente inattaccabile dalle altre sostanze. Nessuno dei due acidi che compongono l'acqua regia riuscirebbe singolarmente ad attaccare l'oro ed il platino; d'altra parte vi sono alcuni metalli particolari, come il tantalio, l'iridio, l'osmio e pochi altri, che sono eroicamente in grado di resistere anche all’acqua regia! Acqua forte: è acido nitrico, HNO3, al 65%; si usava e si usa ancora per la corrosione del rame nella fabbricazione delle cosiddette "acqueforti"; è una tecnica di stampa nella quale lastre di rame sono incise opportunamente con immagini per mezzo dell'acido, e forniscono le matrici per delle vere opere d'arte, alle quali si sono cimentati grandissimi artisti del passato e del presente (allego in fondo come piccolo esempio una vecchia cartolina ottocentesca ottenuta all'acquaforte).
Acqua ragia: dal greco "radnis" goccia, si intende per acqua ragia un distillato ottenuto dalla resina che stilla dalle conifere, specialmente dal pino marittimo. Mi è capitato di vedere di persona questa raccolta qualche anno fa campeggiando in un suggestivo bosco portoghese circondato da queste profumate piante, ognuna col suo barattolino alla base pieno di resina! L'Acqua in sè è il principale solvente della natura, ma ciò è sottinteso e non val la pena di aggiungere nulla a questo concetto, che è fondamentale e complesso dal punto di vista chimico-fisico, ma che esula da questi nostri semplici discorsi. Le mie 16 liquide mascherine del Carnevale Chimico sono sfilate, magari qualcuna mi è anche sfuggita... se la trovate in giro, ditemelo! Mi spiace solo non poter abbinare ad ogni "acqua" una adeguata immagine, che ci sarebbe stata benissimo! Ma ne sarebbe risultato un post troppo ingombrante (lo è già così com'è!) ed allora ne metto solo una in rappresentanza di tutte e in omaggio, se mi è permesso il singolare accostamento, all'acido nitrico e a Durer, con la sua acquaforte sull'Arte e l'Alchimia.
|

|
Post n°88 pubblicato il 14 Marzo 2011 da paoloalbert
In chimica analitica tradizionale s'intende per reazione specifica una reazione (che si evidenzia in qualche modo) che avviene solo con un determinato catione o anione.
Anche questo metodo di analisi è merito di quello spirito intraprendente di Fritz Feigl (1923, foto) del quale già ho parlato brevemente in occasione del periodato di potassio. Materiale occorrente: -soluzione al 5% di benzoinossima in etanolo
Il metodo con la carta da filtro è comunque molto più sensibile e corretto. Un altro reattivo interessante e molto simile che dà più o meno le stesse reazioni con il rame è la salicilaldossima, che da soluzioni debolmente acetiche forma un precipitato giallo-verde. |
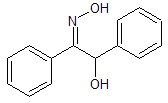
 -Mettere in un piccolo becker qualche ml di
-Mettere in un piccolo becker qualche ml di 
|
Post n°87 pubblicato il 11 Marzo 2011 da paoloalbert
Il 2011 è stato nominato dall'ONU, dall'UNESCO e dalla IUPAC "Anno internazionale della Chimica".
L'ACQUA: UNA SOLUZIONE CHIMICA, è il tema proposto; l'argomento, pur esplicito, non è letteralmente vincolante poichè sono ammesse opportune divagazioni trasversali dalla secca formula H2O.
Apprezzando l'invito di Gifh a partecipare a questa edizione del Carnevale, che ha scopo divulgativo scientifico ed è dedicato principalmente ai non chimici, tenterò di scrivere qualcosina che mi verrà in mente che abbia attinenza al tema proposto. La scadenza del "gioco" è il 23 marzo, quindi se entro tale data si leggerà qualcosa di acquoso sul blog, quella sarà la mia mascherata...
|
|
Post n°86 pubblicato il 08 Marzo 2011 da paoloalbert
Per quella serie di considerazioni aerodinamiche viste, per le quali le pale nella rotazione attorno all'asse del rotore sono alternativamente una forza motrice e un freno, la soluzione migliore (se non ci fossero problemi di bilanciamento) si otterrebbe con un sistema monopala, che avrebbe la maggior velocità di rotazione e quindi maggior rendimento meccanico al sistema di ingranaggi per l'alternatore collegato all'asse. Fondamentale per questi lavori è la legge di Betz che mette in relazione la potenza ottenibile al rotore con i parametri del vento e della superficie descritta dall'elica: P = 0,5 d S v (v12 - v22) Salta immediatamente all'occhio che nella formula non appare il numero delle pale, ma l'area da esse sottesa nella rotazione e che l'energia è proporzionale al cubo della velocità! La rotazione delle pale sul loro asse viene usata per angolarle in modo che presentino un differente angolo di attacco al vento e mantengano costante il loro numero di giri indipendentemente dalla forza vento; quando esso supera i limiti di progettazione le pale vengono messe in bandiera e il sistema si ferma. Il fatto che le pale siano sottili (cioè che abbiano un forte allungamento), è per migliorare il rendimento aerodinamico delle stesse, in quanto in questa configurazione si riducono le resistenze indotte, come nel principio delle ali degli alianti, che hanno allungamenti spaventosamente più elevati rispetto a quelli di un aereo a motore. Da tutte queste lunghissime considerazioni (un discorso a puntate così lungo di sicuro non lo faccio più... ma dovevo prima di tutto essere convinto io stesso!), si deduce che il sistema a tre pale è la soluzione più usata perchè è quella col maggior rendimento fluidodinamico/economico: Spero che anche Guglielmo si sia convinto: io tutto quello che avevo da dire l'ho detto. |
|
Post n°85 pubblicato il 06 Marzo 2011 da paoloalbert
Ora facciamo finta che il nostro generatore eolico abbia solo due pale. Da queste considerazioni si vede chiaramente che quando le pale sono orizzontali rispetto al terreno, la pala che scende diventa un freno per la pala che sale, e viceversa! Proviamo allora ad aggiungere una ulteriore pala e creiamo il rotore a tre pale poste a 120 gradi. Se usiamo un rotore a 4 pale torniamo al problema delle 2 pale ma con doppio peso; se ne usassimo di più (per esempio cinque, sette), per la teoria delle turbine a canale aperto (dove non c'è una voluta di contenimento al flusso del fluido) le resistenze sulle pale dipendono dal numero delle pale stesse, per cui più numerose sono, maggiore è la resistenza al vento e minore è la velocità di rotazione. La prossima puntata aggiungerà alteriori elementi e sarà finalmente quella conclusiva. |
|
Post n°84 pubblicato il 04 Marzo 2011 da paoloalbert
- Ma perchè mai i generatori eolici hanno tutti tre pale?- mi chiese qualche tempo fa il mio amico Guglielmo, anche lui come il sottoscritto un "curiosone" di scienza e tecnica. L'obiezione che il profano (intendo il profano evoluto, non colui che bovinamente non si chiede mai niente!) fa di solito è: Il mio amico giustamente osservava che i rotori eolici usati per pompare l'acqua nelle fattorie western hanno tantissime palette, tante che quasi non ci vede attraverso... quelli sì che dovrebbero rendere! I moderni generatori eolici basano invece il loro funzionamento non sulla spinta ma sull'effetto di portanza creato sulle pale dal flusso del vento; le pale in questo caso non sono più dei pannelli piani ma sono strutture a profilo alare altamente efficenti dal punto di vista aerodinamico, esattaemente come la differenza tra il flusso di aria che scorre sotto e sopra le ali di un aereo lo tiene sollevato o come una barca Hi-Tech che partecipa alla America's Cup vola sull'acqua grazie al flusso d'aria che scorre davanti e dietro alle sue vele. Eliminiamo quindi subito i generatori "tipo western" (la cui efficienza è paragonabile agli ottocenteschi battelli a pale del Mississippi) e teniamo buoni quelli moderni (la cui efficienza è paragonabile alle eliche delle superpetroliere). Fine della prima puntata: poichè il discorso tutto unito era troppo lungo e diventava noioso, lo devo spezzare addirittura in tre parti; fra un po' di tempo la seconda puntata. |
|
Post n°83 pubblicato il 01 Marzo 2011 da paoloalbert
Anche se sono estimatore e goloso consumatore dei buoni olii di oliva italiani, non starò certo a tesserne le lodi in questa sede inappropriata; essa si presta ad "assaggiarlo", ma solo dal punto di vista chimico... estere + acqua --> acido + alcol, quindi: Questa parziale idrolisi si esprime analiticamente in quantità di acido oleico libero, ed è in pratica l'acidità dell'olio, quella che quando è eccessiva lascia in gola una spiacevole sensazione di "ruvidità". Si dice anche che l'olio acido "raschia in gola". Per pura curiosità avevo controllato tempo fa un campione di rinomato olio extravergine delle colline moreniche del Garda, commercialmente definito "a bassissima acidità"; recentemente per questo lavoro ho rifatto il test su un altro ottimo olio ma volutamente più "stagionato" (era da qualche mese in bottiglia chiusa ma non piena) andando a verificare, con un margine di errore tollerabile, le rispettive acidità. Materiale occorrente: - miscela 1:1 etanolo/etere resa perfettamente neutra alla fenolftaleina con qualche goccia di idrossido di potassio KOH A 5 g esatti di olio vengono aggiunti in una beuta 50 ml della miscela alcool/etere, agitando fino a soluzione.
Se n è il volume in ml consumato, l'acidità in oleico è espressa dalla formula: % oleico = n x 0,282 ml di KOH consumati nelle due prove: 0,95 e 2,6 L'olio extravergine dovrebbe avere acidità inferiore all'1%; nel campione analizzato in precedenza avevo ottenuto un valore veramente basso di 0,27%, quindi soddisfacente pienamente i requisiti dichiarati, sia dal punto di vista organolettico (il più importante!) che da quello chimico. |


|
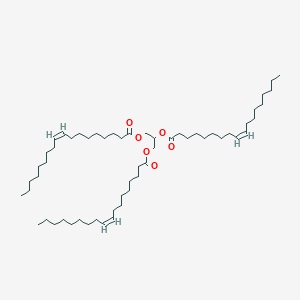
Post n°82 pubblicato il 23 Febbraio 2011 da paoloalbert
Chimicamente, di cosa è fatto l'olio di oliva? -glicerina: HO-CH2-CH(OH)-CH2-OH è un alcol trivalente, tre sono gli ossidrili -OH a disposizione per essere esterificati dagli acidi; ricordiamo che ad ogni ossidrile "si attacca" una molecola di acido eliminando una molecola di acqua. -acido oleico: CH3-(CH2)7-CH=CH-(CH2)7-COOH -acido palmitico: CH3-(CH2)14-COOH -acido linoleico: -acido stearico: CH3-(CH2)16-COOH Li ho elencati in ordine di abbondanza, tralasciando gli altri acidi presenti in quantità molto bassa (palmitoleico, linolenico, arachico, eicosenoico, beenico) e considerando sottinteso olio extravergine di oliva. L'acido oleico è presente in percentuale minima del 65%, ma supera di solito il 75%
Quei serpentelli rappresentano la catena di atomi di carbonio (dove c'è un angolo lì c'è un -CH2-) ed il nucleo centrale è la glicerina salificata con tre atomi di acido oleico. Osservare anche che due acidi (oleico e linoleico) hanno nella molecola uno e due doppi legami, il chè fa prendere il nome di "insaturi" a questi acidi e danno pregio al nostro ottimo grasso vegetale per le sue capacità antiossidanti (assieme ai polifenoli), oggi giustamente tanto valorizzate; per confronto, l'acido principale presente nel burro è il palmitico, che è invece un acido saturo (senza doppi legami, ved. sopra). Ma allora, tutto ciò premesso, l'olio è acido o non è acido, visto che (scusate il bisticcio di parole) gli acidi sono tutti esterificati e quindi non presenti fisicamente nel giallo e profumato liquido? |
|
Post n°81 pubblicato il 19 Febbraio 2011 da paoloalbert
No, gli esteri non sono coloro che vivono oltre confine... Nel post 56 s'era parlato di acidi (quelle sostanze che se fossero auto avrebbero tutte una targa con su scritto --> -COOH) Chiamiamo R- tutta la parte di molecola dell'acido che non sia -COOH
R-COOH + R'-OH --> R-COO-R' + H2O Quello che si è formato, R-CO-OR' è un estere! Naturalmente per far avvenire la reazione dobbiamo mettere in atto una procedura idonea al caso, per esempio con un catalizzatore acido (H+ sopra la freccia), giusta temperatura e tempo di reazione e tener presente che la reazione stessa è sempre di equilibrio, cioè la freccia non va esclusivamente verso destra ma a seconda delle condizioni di lavoro anche verso sinistra, creandosi un equilibrio fra reagenti e prodotti. Prendiamo per esempio due sostanze da tutti conosciute, l'acido acetico e l'alcol etilico, le cui formule ormai sono note: Ecco la reazione, messa senza il complesso meccanismo, proprio terra-terra: CH3-COOH + HO-CH2-CH3 <--> CH3-COO-CH2-CH3 + H2O Acido ed alcol si sono abbracciati ciascuno con la proprio parte "sensibile" (rispettivamente il carbossile e l'ossidrile), alla fine si è eliminata una molecola d'acqua (OH dall'acido + H dall'alcol = H2O) e si è formata una nuova sostanza di nome... acetato di etile! Anche nel nome il processo ricorda la salificazione.
Gli esteri sono diffusissimi in natura e quelli a pochi atomi di carbonio sono liquidi volatili dal caratteristico profumo, tant'è che vengono detti "esteri di frutta" per il loro piacevole aroma che per qualcuno di essi ricorda proprio l'odore di frutti maturi. |
 Il padre delle esterificazioni secondo questo metodo (ce ne sono altri!) è il grande chimico tedesco premio Nobel
Il padre delle esterificazioni secondo questo metodo (ce ne sono altri!) è il grande chimico tedesco premio Nobel |
Post n°80 pubblicato il 15 Febbraio 2011 da paoloalbert
Nel post n.56 avevo dedicato una puntata di queste mie riflessioni a quegli occasionali passanti ai quali non sarebbe dispiaciuto sentirsi dire senza tante complicazioni cosa sono gli acidi carbossilici. In principio era l'Idrocarburo... si potrebbe dire, e devo per forza partire da qui, facendomi scudo con mille sottintesi omissis... l'etano H3C-CH3 è un alcano; l'etilene H2C=CH2 è un alchene; l'acetilene H-C≡C-H è un alchino. Ora se da un idrocarburo eliminiamo un atomo di idrogeno e lo sostituiamo con un gruppo ossidrile -OH, otteniamo un'altra classe di composti, completamente diversa in tutto e per tutto dai genitori di origine: gli alcoli, che acquistano nel nome il suffisso "olo". CH3-H cioè il metano, dà origine a CH3-OH, l'alcool metilico o detto più propriamente metanolo. E se in un idrocarburo sostituiamo due idrogeni con due ossidrili? E' possibile questo? Certo! Ne derivano gli alcoli diossidrilici, triossidrolici... poliossidrilici, ognuno col suo bel nome e le sue interessanti proprietà. Ci sarebbero da aggiungere un'infinità di cose sugli alcoli, ma termino qui per non approffittare della pazienza dei volonterosi. |
|
Post n°79 pubblicato il 12 Febbraio 2011 da paoloalbert
Nel ragionamento sulle coppie del post 77 si parlava di solubilità (o meglio, insolubilità) dei sali, che è stato poi messo in pratica con la preparazione del bromato di bario nel post 78. Si potrebbe pensare che, per quanto restio a sciogliersi, un sale sarà sempre un pochino solubile... non è concepibile che non se ne sciolga nemmeno una molecola in un oceano d'acqua! Ma cosa succede se metto in acqua un sale "del tutto" insolubile? Il prodotto tra le concentrazioni ioniche [Cu++] e [S--] (le concentrazioni si indicano tra parentesi quadre) si chiama Prodotto di solubilità, si indica con Kps ed è una costante che dipende dalla sostanza in esame. Che senso ha un numero così piccolo? Per noi che siamo sperimentatori e vogliamo toccar con mano le cose, se buttiamo una spatolata di CuS in un litro d'acqua quanto se ne scioglierà, in pratica? Kps = [Cu2+][S2-] Kps = 6x10-37 Poichè una molecola di CuS dà origine ad uno ione Cu2+ ed uno solfuro S2- (in quantità identiche), ed il loro prodotto è 6x10-37, per sapere la concentrazione del CuS basterà estrarre (in questo caso) la radice quadrata del Kps, che vale 7,7x10-19 7,7x10-19 x 95 = 7,3x10-17 grammi sciolti in un litro Traducendo terra-terra con un po' di conversioni: per sciogliere completamente un milligrammo di solfuro di rame ci vuole qualcosa come tredici miliardi di metri cubi d'acqua...!!! C'era la sorpresa vero? Ma il CuS non è il più insolubile dei sali! Il solfuro di mercurio HgS (il cinabro) ha un pazzesco Kps di 2x10-53...!!! |
|
Post n°78 pubblicato il 07 Febbraio 2011 da paoloalbert
Per capire bene la preparazione di questo sale da parte dei non chimici, bisognerebbe leggere la storiella dell'altra volta, riguardo lo scambio di coppie. Per prepararlo ho usato prorio una reazione di metatesi, facendo scambiare tra di loro due metalli (bario e potassio) con altrettanti "radicali acidi" (cloruro e bromato). La reazione è la seguente: BaCl2 + 2 KBrO3 --> 2 KCl + Ba(BrO3)2 All'inizio abbiamo cloruro di bario e bromato di potassio; alla fine avremo cloruro di potassio e bromato di bario... un bello scambio di coppia! Procedura: - in un becker da 50 ml sciogliere 10 g KBrO3 in 21 ml di acqua bollente ed in un altro becker sciogliere 6,2 g BaCl2 in 8 ml di acqua bollente. Attenzione che l'acqua è in quantità proprio al limite della solubilità dei sali a 100°, pertanto i sali stessi non si scioglieranno completamente fino al raggiungimento del punto di ebollizione. Se del caso aggiungere qualche goccia d'acqua fino a soluzione limpida.
Il bromato di bario è un sale dalle interessanti proprietà ossidanti; si decompone per esempio volentieri sviluppando una bella fiamma verde, come quella che vediamo durante gli spettacoli pirotecnici, dei quali abbiamo già parlato.
|

|
Post n°77 pubblicato il 02 Febbraio 2011 da paoloalbert
Non fraintendiamo, si parla di coppie... chimiche! Allora torniamo alle nostre coppie umane, Aldo con Bruna, Carlo con Daria. Riassumendo: A+B- + C+D- (in acqua) --> A+ + B- + C+ + D- Ma cosa succede se un maschio ed una femmina (mettiamo che siano Carlo e Bruna) che prima non stavano insieme ed ora si trovano liberi di ballare al buio, hanno un terribile colpo di fulmine l'un l'altra? Riassumendo AB + CD --> AD + CB (e CB se ne vanno per conto loro) Si sono formate due nuove coppie per "doppio scambio", solo perchè i due nuovi partner CB sono fuggiti assieme! Esempio: avverrà la reazione tra cloruro di ferro e solfato di zinco per formare cloruro di zinco e solfato di ferro? FeCl2 + ZnSO4 --> FeSO4 + ZnCl2 ? Dobbiamo chiederci: si formarà un sale insolubile? Risposta: no, sia il cloruro di zinco che il solfato di ferro sono solubilissimi in acqua e rimangono disciolti sotto forma di ioni in equilibrio tra di loro. La reazione non avviene. Sostituiamo solo il ferro col calcio: cloruro di calcio e solfato di zinco formeranno cloruro di zinco e solfato di calcio? CaCl2 + ZnSO4 --> CaSO4 + ZnCl2 ? Siccome il solfato di calcio CaSO4 è insolubile, precipita (si dice così!) sul fondo del recipiente e si separa: la reazione di doppio scambio avviene, perchè la nuova coppia calcio/solfato è fuggita, lasciando da soli gli altri due! Sembra incredibile, ma questo semplice concetto di swapping tra le coppie rimane fonte di dubbio fino alla maturità ed oltre... |
|
Post n°76 pubblicato il 28 Gennaio 2011 da paoloalbert
Ambientandoli in periodo vittoriano, ho parlato poco tempo fa di certi singolari avvelenamenti il cui colpevole era il verde di Parigi. Chimicamente questo composto colorante è un sale di rame e sappiamo che quasi tutti i sali di rame o sono azzurri o sono verdi. Un pigmento per essere utilizzabile deve essere del tutto insolubile in acqua e per quanto possibile inalterabile nel tempo, quindi in pratica i sali di rame effettivamente adatti allo scopo sono molto pochi. Molto simile al verde di Parigi è il verde di Scheele, al quale manca però la componente acetica ed è pertanto arsenito di rame CuHAsO3; anch'esso non ha sempre una ben definita composizione poichè i rapporti tra arsenico, rame e molecole d'acqua legate dipendono dal metodo di preparazione. In ambito artistico era uno dei verdi preferiti da Cezanne, Van Gogh e altri artisti di quella splendida età pittorica che fu l'impressionismo.
Lo accompagno volentieri con un quadro di Cezanne e uno di Monet, nei quali, ci giurerei, l'acetoarsenito di rame è il protagonista fondamentale. E poi dicono che la Chimica e l'Arte non vanno d'accordo...
|



|
Post n°75 pubblicato il 23 Gennaio 2011 da paoloalbert
In un laboratorio di chimica sperimentale bene organizzato, i reagenti (quelli che le persone comuni chiamano "le sostanze"), dovrebbero essere talmente numerosi da rasentare l'assurdo, per permettere (almeno in teoria) di progettare una qualsiasi reazione che capitasse di dover fare.
Materiale necessario: - Potassio nitrato KNO3 Stavolta occorre fare una sottolineatura sul termine "opportuna": la vetreria deve essere categoricamente "normalizzata", ovvero di qualità e con tutti i giunti di vetro smerigliato di dimensioni standard, per assicurare una tenuta perfetta; niente deve essere di gomma, plastica o materiali non resistenti alla potentissima aggressività dell'acido nitrico concentrato. La semplice reazione da sfruttare sarà la seguente: KNO3 + H2SO4 --> KHSO4 + HNO3 - In un pallone da 500 ml introdurre 100 g di KNO3 e 100 ml (184 g) di H2SO4 concentrato. L'acido solforico è in grande eccesso (quasi il doppio) rispetto alla quantità stechiometrica e serve per trattenere il più possibile l'acqua derivante dalla decompozione dell'acido nitrico. Agitare brevemente il pallone e chiudere immediatamente predisponendo il tutto per la distillazione, con adatto termometro e refrigerante Liebig.
Il peso di 10 ml esatti è stato di 15 g, quindi l'acido ottenuto in questo modo ha densità 1,5 - corrispondente ad una concentrazione del 96%, esattamente come mi ero proposto di ottenere.
Quest'acido mi è poi servito per la nitrazione dell'anidride ftalica (ved. luminol); il rimanente servirà per altre nitrazioni toste, nei casi in cui è difficile introdurre un nitrogruppo -NO2 in una molecola se non si ha a disposizione HNO3 quasi al 100%.
|


 Tutto deve potere essere svolto con la massima tranquillità e sicurezza.
Tutto deve potere essere svolto con la massima tranquillità e sicurezza.
|
Post n°74 pubblicato il 16 Gennaio 2011 da paoloalbert
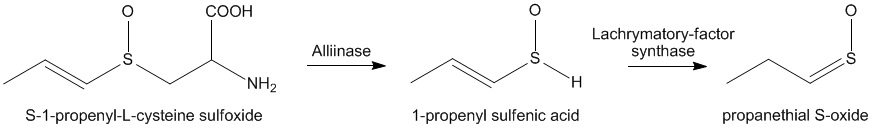
In questo blog capita talvolta uno strampalato "chef" chimico che va a preparare le sue indigeste ricette (magari a base di 2-naftalensulfonato sodico o peggio!) e poi tenta di propinarle a quei benevoli commensali che si prestano agli esperimenti, non so con quanto successo... Ecco, e te pareva! dirà qualcuno, vedrai che ricadiamo inesorabilmente nella chimica e adesso salterà fuori la storia del perchè la cipolla fa piangere chi la taglia. Allora, veniamo al sodo, perchè si piange? (Spero sia sottinteso che non si intende "zolfo" in quanto elemento, ma che esso è combinato chimicamente a formare sostanze con caratteristiche peculiari che con l'elemento non hanno niente a che vedere. Ma quella particolare molecola di cui dicevo non è sufficiente: ci vuole qualcos'altro, tipicamente biologico.
Successivamente questa sostanza per rapido riarrangiamento (riarrangiamento si ha quando in una molecola cambiano di posto certi atomi o gruppi di atomi rispetto alla posizione di partenza) porta alla formazione di... (rullino i tamburi): Questa tioaldeide è un gas reattivo e irritante, che stimolando i neuroni sensoriali degli occhi agisce come un agente di lacrimazione e di bruciore, al quale reagiscono le ghiandole lacrimali generando liquido allo scopo di tentare di diluire il fastidioso agente chimico estraneo. Ci sono vari e più o meno fantasiosi consigli per cercare di evitare questo piccolo inconveniente a cui va incontro ogni brava massaia alle prese con un soffritto di cipolla; sono consigli che magari si provano una volta ma che poi nessuno per fretta e semplicità mette in pratica. Quindi vai tranquillo cuoco: zac-zac-zac, una bella affettatina senza starci proprio sopra con gli occhi, olio quanto basta, uno spicchietto d'aglio e un rametto di rosmarino... e lasciamo pure che il povero propanetiale sulfossido tenti anche lui di fare il lavoro di difesa per il quale è stato creato. |
Inviato da: paoloalbert
il 02/05/2021 alle 21:53
Inviato da: Bepi1249
il 15/04/2021 alle 14:11
Inviato da: Tanner85
il 06/02/2021 alle 00:01
Inviato da: poetryclub
il 04/10/2020 alle 23:00
Inviato da: poetryclub
il 04/10/2020 alle 20:58