CHIMICA sperimentale
Esperienze in home-lab: considerazioni di chimica sperimentale e altro
|
Post n°53 pubblicato il 30 Ottobre 2010 da paoloalbert
Oggi propongo fast food, un piatto di chimica secca e veloce, niente commenti, niente divagazioni!
Materiale occorrente:
Il residuo della distillazione è più altobollente e leggermente giallino ed ha quasi identico profumo. |
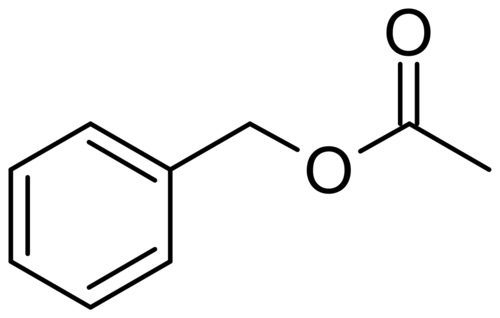
 - In un pallone da 250 ml introdurre
- In un pallone da 250 ml introdurre 
|
Post n°52 pubblicato il 27 Ottobre 2010 da paoloalbert
Fra i molti metodi di ricerca analitica del rame c'è una curiosa e tanto sensibile procedura, che perfino il Treadwell (la Bibbia della chimica analitica classica) afferma: "...è questa una reazione troppo sensibile per le normali ricerche analitiche"! Oggi lo chef va a preparare due soluzioni:
|
|
Post n°51 pubblicato il 24 Ottobre 2010 da paoloalbert
Chimico austriaco ebreo, riuscito durante le persecuzioni naziste a rifugiarsi in Brasile, Fritz Feigl è considerato il padre delle "analisi al tocco", ovvero di quelle semplici procedure analitiche svolte senza bisogno di sofisticate apparecchiature ma solo di una piastrina di porcellana (o addirittura carta) e di qualche opportuno reattivo. Avendo preparato il periodato di potassio KIO4 (della cui sintesi parlerò prossimamente), ho voluto verificare questo test, per ora solo qualitativamente e senza provarne la sensibilità; nella fotografia finale si può vedere il risultato. conseguente colorazione viola intensa caratteristica. 2 Mn++ + 5 IO4- + 3 H2O -> 2MnO4- + 5 IO3- + 6 H+ Naturalmente il saggio può avvenire anche all'opposto, per rivelare tracce di manganese per mezzo del periodato, come probabilmente avveniva in Amazzonia. Si opera in questo modo:
|

|
Post n°50 pubblicato il 18 Ottobre 2010 da paoloalbert
Navigando nel gran mare della Rete ci si imbatte talvolta in informazioni curiose ed interessanti; succede di solito quando si cerca tutt'altro e andando ad aprire pagine dopo pagine come scatole cinesi, vengono a galla notizie insospettate. ________________________________________ |
|
Post n°49 pubblicato il 14 Ottobre 2010 da paoloalbert
Come annunciato, ecco un saggio del contenuto del libro dell'Ambrosioni, nel quale si parla del solfato di rame, del modo e dei pochi luoghi di produzione. Ecco il testo originale: -"Anche questo Sale porta in commercio varj nomi, i quali sono: Coparosa, Pietra, o Vetriolo Turchino, Vetriolo di Cipro, di Rame, o di Venere.
|
|
Post n°48 pubblicato il 13 Ottobre 2010 da paoloalbert
Sicuramente uno dei libri più preziosi che ho nella mia piccola biblioteca è un manuale indue volumi di non grande formato ma di circa 300 pagine l'uno: l'autore è il sig. Felice Ambrosioni, il titolo è "Manuale per i Droghieri" ed è edito in Pavia nel 1823. La consistenza e l'odore della cellulosa, le macchie marroni proprio sul titolo, il delizioso color giallino della carta, tutto esprime il lungo tempo passato e l'uso intenso che una volta si faceva dei libri, oggetti troppo preziosi per non essere sfogliati e risfogliati più volte, consultati, letti insomma come ogni libro serio impone!
Ho trovato sulla rivista Caleidoscopio Letterario, trattando della Chimica Clinica Italiana dell'Ottocento, una ottima biografia del nostro Ambrosioni, che brevemente riassumo.
|

|
Post n°47 pubblicato il 09 Ottobre 2010 da paoloalbert
Se è vero che non tutte le ciambelle riescono col buco, altrettanto si può dire che non tutte le sintesi... riescono!
|
 Ho cercato di fare tutto come si deve (grande eccesso di uno dei componenti, temperatura controllata, tempo ab abundantiam, ecc.), ottenendo però alla fine, dopo l'estrazione con etere, un prodotto resinoso marroncino, estremamente appicicaticcio e fastidioso, di odore balsamico ma nemmeno lontanamente cristallizzabile e men che meno degno di prendere il nome di
Ho cercato di fare tutto come si deve (grande eccesso di uno dei componenti, temperatura controllata, tempo ab abundantiam, ecc.), ottenendo però alla fine, dopo l'estrazione con etere, un prodotto resinoso marroncino, estremamente appicicaticcio e fastidioso, di odore balsamico ma nemmeno lontanamente cristallizzabile e men che meno degno di prendere il nome di 
|
Post n°46 pubblicato il 04 Ottobre 2010 da paoloalbert
L'altra volta abbiamo preparato l'acido cinnamico: che ce ne facciamo? Andiamo subito a sacrificarne un po' per una nobile causa! La sintesi di oggi è dedicata agli appassionati (come il sottoscritto) degli esteri odorosi, e produrrà un buon composto, un estere proprio da non perdere! Materiale occorrente:
- In un palloncino da 100 ml introdurre 12 g di acido cinnamico e 33 ml di metanolo; aggiungere lentamente agitando 1,5 ml di H2SO4 conc.- Predisporre per il riscaldamento a ricadere con il sistema più opportuno (io uso il bagno a sabbia) e mantenere a lento riflusso per cinque ore.
|

 Si ottiene il
Si ottiene il 
|
Post n°45 pubblicato il 29 Settembre 2010 da paoloalbert
In un laboratorio chimico, anche a livello hobbistico come il mio, il nitrato d'argento AgNO3 è uno dei reagenti fondamentali che non può mancare (ed infatti non manca!) anche se ne è abbastanza costoso l'acquisto; in caso di necessità è possibile prepararselo da soli con un facile procedimento elettrochimico, avendo a disposizione un oggetto commerciale "d'argento", ovvero una lega di argento e rame. La procedura seguente vale per qualsiasi altro rottame d'argento o comunque cose meno nobili della bellissima moneta testimone del boom economico italiano; naturalmente il valore affettivo dell'oggetto non dovrà superare quello venale del vil metallo che si andrà ad estrarre... Ecco come ho proceduto, punto per punto: - Pesare l'oggetto, nel mio caso 11 g - Metterlo in un becker con 10 ml di acqua e aggiungere 20 ml di acido nitrico (HNO3) concentrato - Scaldare un po' per avviare la reazione, dopo di chè essa procede vigorosamente da sola, con abbondante svolgimento di ossidi d'azoto (NO2) - A dissoluzione avvenuta (il liquido diventa azzurro) diluire portando a 50 ml con acqua - Neutralizzare con precisione l'eccesso di acido con la quantità quasi strettamente necessaria di sodio bicarbonato (NaHCO3), mescolando bene ad ogni piccola aggiunta fino a svolgimento completo della CO2 - Non arrivare a pH alcalino (altrimenti i due metalli precipitano come ossido e carbonato) ma tenerlo appena acido, la soluzione deve sempre essere azzurra e limpida - Porre la soluzione dei nitrati AgNO3/Cu(NO3)2 in un cilindro graduato da 50 ml (un recipiente alto e stretto facilita le cose) - Pesare una laminetta di rame lunga e stretta e ben pulita con HNO3 diluito e poi risciacquata (nel mio caso 12,1 g) ed immergerla nel cilindro, appendendola al bordo superiore del cilindro stesso - Si nota immediatamente una copiosa "nevicata" di argento metallico che si stacca dal rame e precipita sul fondo del cilindro (potenziale elettrochimico del rame +0,34 V, quello dell'argento +0,80 V) - Ogni tanto dare una mescolata, lasciar decantare e riimmergere la laminetta fino a precipitazione completa (occorrono solo un paio d'ore, l'operazione è veloce) - Sincerarsi che alla fine il liquido non contenga più ioni argento ma solo rame (fare un test con HCl, non deve precipitare l'AgCl) - Filtrare il precipitato grigio metallico e lavarlo abbondantemente con acqua fino a scomparsa totale del rame - Lasciar asciugare bene all'aria la polvere metallica, pesarla e riporla poi ben chiusa al riparo dall'aria; la quantità di Ag ottenuta è stata nel mio caso di 8,9 g Ecco anche un po' di stechiometria (ometto i calcoli perchè sono banali): 3 Ag + 4 HNO3 --> 3 AgNO3 + NO +2 H2O 2 AgNO3 + Cu --> 2 Ag + Cu(NO3)2 Partendo da 11 g ed ottenendo 8,9 g di Ag, la % di questo metallo nella moneta è stata pari al 81%, il resto della lega è rame o poche altre impurezze che non ci interessano.
|

|
Post n°44 pubblicato il 27 Settembre 2010 da paoloalbert
Niente reazioni chimiche stavolta. Ho trovato (attenzione, trovato, non inventato...) un ingegnoso sistema, fisico-matematico, per riuscire a conoscere la composizione di una lega di due metalli (noti) ma di ignota percentuale relativa. a- Se ne pesa esattamente un frammento non tanto piccolo (approssimazione almeno 0,1 g) Inutile dire che la precisione nei risultati dipende essenzialmente da questa misura, da eseguirsi con l'aiuto di una buona buretta graduata. Si pongono allora in definitiva i seguenti dati: x = peso primo metallo in g Si fa ora questo semplice sistema matematico: {x + y = P risolto il quale in x e y si hanno i pesi e quindi le % relative dei due metalli! Capito niente? Nessun problema, facciamo un esempio pratico. Proviamo con una targhetta in ottone (sappiamo che è fatta di rame + zinco); ma quanto rame e quanto zinco? Peso = 34,8 g {x + y = 34,8 risolvendo si trova: x = 27,2 g e y = 7,6 g Adesso sembra semplice, vero? (E se la lega fosse ternaria? |
|
Post n°43 pubblicato il 23 Settembre 2010 da paoloalbert
Nel 1868 William Henry Perkin scoprì una reazione delle aldeidi aromatiche che è preziosa per la sintesi degli acidi a,ß-insaturi: se si fa condensare una aldeide aromatica con l'anidride di un acido alifatico e col sale alcalino dello stesso acido, si ottiene appunto un acido insaturo, e questa reazione ha preso il nome dal grande chimico inglese, suo scopritore.
Ecco quanto occorre per la sintesi abbastanza laboriosa di questo interessante acido, la cui corrispondente aldeide (aldeide cinnamica C6H5-CH=CH-CHO) costituisce il componente principale dell'aroma della cannella: Prima della sintesi vera e propria va preparato l'acetato di potassio anidro, riscaldando a 150° in forno il sale normale triidrato. Esso prima fonde nella sua acqua di cristallizzazione e poi lentamente anidrifica; lasciarlo in forno almeno tre ore e poi velocemente polverizzarlo ancora caldo in un mortaio e conservarlo ermeticamente chiuso.
Si separa del solido ed anche a pH alcalino rimarrà insolubile una parte gialla leggera resinosa galleggiante, da eliminare pazientemente per decantazione o pipettando il liquido, sempre quasi all'ebollizione. La resa ne risente, ma si evita la decolorazione con carbone animale ed il prodotto finale è quasi bianco e puro. Inutile filtrare in questa fase perchè per raffreddamento si intasano immediatamente filtro e imbuto.
Si deve ottenere (a caldo, eventualmente aggiungere acqua fino ad un volume totale di circa 500 ml) un liquido limpido solo leggermente giallino. Ecco quì sotto l'acido cinnamico che si sta tranquillamente asciugando al sole.
|
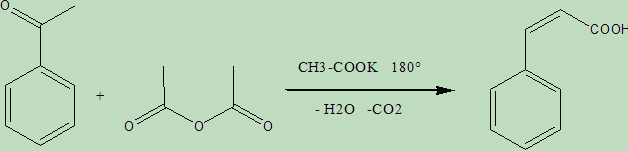
 - In un pallone da 250 ml con refrigerante a ricadere e tappo a CaCl2 si introducono
- In un pallone da 250 ml con refrigerante a ricadere e tappo a CaCl2 si introducono  Continuare ad aggiungere
Continuare ad aggiungere 

|
Post n°42 pubblicato il 03 Settembre 2010 da paoloalbert
Leggendo l'interessante volume "Molecules that changed the World" di Nicolau-Montagnon, si trova ad un certo punto la storia della sintesi della canfora, effettuata dal finlandese Gustaf Komppa nel 1903. Ci stanno sedici provette (anche troppe!) di quelle grandi e buone, da 180x18 della Schott-Duran, in due file da otto, una fila bassa davanti ed una alta dietro, di una comodità estrema, come il sedile di una vecchia carrozza.
Per le foto ho messo qualche provetta con in ciascuna un liquido colorato per fare un po' di scena, come si usa per i liquidi "di chimica" (nei film in cui appare un laboratorio di chimica c'è sempre il pallone viola col permanganato, quello giallo col cromato e quello azzurro col solfato di rame...).
|
 Finalmente un portaprovette degno del nome, che sta bello dritto e stabile, non ha il tramezzo centrale sempre incurvato, non si offende se si trova ad una spanna dal bunsen ed ha una linea old style come piace a me.
Finalmente un portaprovette degno del nome, che sta bello dritto e stabile, non ha il tramezzo centrale sempre incurvato, non si offende se si trova ad una spanna dal bunsen ed ha una linea old style come piace a me.
|
Post n°41 pubblicato il 26 Agosto 2010 da paoloalbert
Buttare un vecchio Hard-disk? Non sia mai! Ci sono dentro delle piccole ma potentissime calamite che per essere così potenti sono al ferro-boro-neodimio: non potrebbero essere quindi per un chimico sperimentale una perfetta miniera d'oro... pardon, di neodimio? Ho ripetuto un paio di volte l'operazione fino ad ottenere una soluzione perefttamente limpida (di colore vagamente violetto pallido) e solo leggermente acida.
Partendo da un magnete di 6 g, dopo tutti i passaggi, i lavaggi e le inevitabili perdite, ho ottenuto alla fine 1,7 g di NdF3, che ritengo adeguatamente puro per lo scopo di questo lavoro di ludica curiosità sperimentale. E il fiorellino si era messo proprio lì dove di solito faccio le fotografie, forse per smentire con poesia coloro che tentano (purtroppo con successo) perfino di rendere arida questa scienza meravigliosa.
|
 Ora entro nel merito in maniera concreta: la lega al ferro-boro-neodimio di composizione indefinita di cui sono fatti questi magneti si scioglie velocemente e perfettamente in
Ora entro nel merito in maniera concreta: la lega al ferro-boro-neodimio di composizione indefinita di cui sono fatti questi magneti si scioglie velocemente e perfettamente in 
 Essendo il
Essendo il 
|
Post n°40 pubblicato il 12 Agosto 2010 da paoloalbert
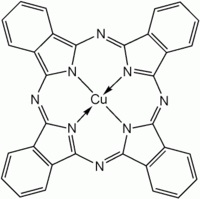
Un pigmento usato per la sua elevatissima stabilità (resiste agli acidi, alle basi, ai solventi, alla luce... a tutto!), non tossico, non inquinante, con in sovrappiù una gran bella molecola da vedere, è la Cuproftalocianina.
Materiale occorrente - Macinare finemente 28 g di urea, 5 g di anidride ftalica e 1,2 g di CuCl2.2H2O; aggiungere circa 50 mg (una puntina di spatola) di molibdato ammonico (NH4)2MoO4 come catalizzatore e mescolare. Porre la miscela in un becher (o ancora meglio in una capsula di porcellana molto grande) e riscaldare per 2-3 ore a bagno d’olio o sabbia, tenendo la temperatura a 180° e mescolando spesso, magari con il bulbo di un termometro (attenzione, non sarebbe la modalità corretta!).
La mia resa è stata di 3,6 g (se ne perde un po' nelle varie operazioni).
|
 La miscela, all'inizio verde, fonde e e schiumeggia e si mantiene a circa 130°, diventando improvvisamente di colore blù; successivamente lo schiumeggiamento aumenta (aumentando molto il volume, attenzione alle fuoriuscite dal recipiente), la T° sale fino ai 180° ed il liquido si trasforma in una pastella violacea densa e appiccicosa, difficile da mescolare.
La miscela, all'inizio verde, fonde e e schiumeggia e si mantiene a circa 130°, diventando improvvisamente di colore blù; successivamente lo schiumeggiamento aumenta (aumentando molto il volume, attenzione alle fuoriuscite dal recipiente), la T° sale fino ai 180° ed il liquido si trasforma in una pastella violacea densa e appiccicosa, difficile da mescolare. Questa fase è fastidiosa perchè bisogna continuare a mescolare (almeno ogni pochi minuti!) quindi occorre armarsi di pazienza ed aver qualcosa da fare nelle immediate vicinanze.
Questa fase è fastidiosa perchè bisogna continuare a mescolare (almeno ogni pochi minuti!) quindi occorre armarsi di pazienza ed aver qualcosa da fare nelle immediate vicinanze.  Dopo raffreddamento, alla massa (viola con riflessi metallici) si aggiungono 150 ml di
Dopo raffreddamento, alla massa (viola con riflessi metallici) si aggiungono 150 ml di 
|
Post n°39 pubblicato il 18 Luglio 2010 da paoloalbert
|




|
Post n°38 pubblicato il 18 Luglio 2010 da paoloalbert
Ma come è fatto il tuo lab?... mi chiede qualcuno... eccolo, in quattro foto nel successivo post n.39! -Il visitatore occasionale vede perciò il mio lab spaziando lo sguardo fra le boccettine, ma non cogliendo l'enorme differenza che vi è tra una e l'altra: cloruro di magnesio o anidride acetica sono per lui più o meno la stessa cosa, cioè due ignote sostanze, quasi sicuramente "tossiche" e "corrosive". -Il visitatore evoluto coglie già la differenza fra alcune sostanze, sa la differenza tra chimica organica e inorganica (ma c'è sempre qualche dubbio...), intuisce che quei tubi sono i refrigeranti per distillare, sa che il sodio fa la fiamma gialla e lo stronzio rossa, con lui si può ragionare su un esagono con delle lineette parallele ai lati chiamandolo anello benzenico senza tanto scandalo. -Il visitatore professionale (non ne ho mai avuti nel mio lab, sono talmente rari i chimici...) noterebbe immediatamente la pochezza e la modestia del lab stesso: vedrebbe con moderno terrore che non c'è neanche un apparecchio "con la spina"! (Ovvero uno qualsiasi degli apparecchi elettronici che corredano tutti i laboratori chimici di adesso). Quest'ultimo visitatore, abituato a lab professionali o universitari, ha perfettamente ragione, il suo discorso non fa una piega, la chimica moderna è e deve essere come dice lui! Punti di vista... |
|
Post n°37 pubblicato il 09 Luglio 2010 da paoloalbert
Cosa succede quando "un-fuori-di-testa" per la chimica sperimentale va in vacanza dove non ha il collegamento Internet ma in compenso ha un home-lab a disposizione? Ecco perchè il blog sembra fermo, ma niente paura, reazioni strane covano sempre sotto la cenere... Buona estate 2010 a tutti coloro che per sbaglio o volontà passano da queste parti! |
|
Post n°36 pubblicato il 24 Giugno 2010 da paoloalbert
Eccoci finalmente al traguardo! Il Luminol è pronto e non resta che provare se la lunga sintesi in tre step ci ripaga con qualche fotone... (Ved. bibliografia in Rete) Tutti sanno che il luminol è utilizzato dagli investigatori sulla scena del crimine per individuare anche tracce minime di sangue. Preparare la soluzione A e la soluzione B (le dosi sono puramente indicative, l'esperimento funziona sempre): - la soluzione A è fatta sciogliendo circa 100 mg di Luminol in 3 ml di NaOH al 10% e portata poi a 150 ml con acqua - preparare una beuta da 250 ml contenente la soluzione A e, oscurata la stanza, aggiungere velocemente la soluzione B... godendosi lo spettacolo... mentre la compagnia stupisce! Unico inconveniente: l'effetto intenso dura solo qualche decina di secondi, ma non si può aver tutto! La luminosità gradualmente decrescente persiste invece al buio completo molto più a lungo. Ecco due immagini dell'esperimento:
Enjoy! |


|
Post n°35 pubblicato il 17 Giugno 2010 da paoloalbert
Eccoci giunti in vista del traguardo; se siamo arrivati fin qui abbiamo buone probabilità di vedere la bellissima luminescenza azzurra di questa sostanza! Una volta in possesso del 5-nitro-2,3-diidro-1,4-ftalazinadione (ved. sintesi nella Fase 2) occorre ridurre il gruppo nitrico -NO2 di questo a gruppo amminico -NH2, con la procedura seguente:
La soluzione si riscalda. Dopo che la reazione si è calmata spontaneamente, bollire leggermente per qualche minuto e filtrare velocemente a caldo.
Il 5-amino-2,3-diidro-1,4-ftalazinadione si presenta come una polvere microcristallina gialla ed è sufficientemente puro per gli usi che ne vorremo fare; si potrebbe purificare ulteriormente perdendone un po' ma non lo ritengo necessario per gli scopi essenzialmente "ludici" che mi sono prefisso.
Nella quarta e ultima fase vedremo se questa intrigante sostanza avrà mantenuto le promesse per cui è stata laboriosamente sintetizzata. |
 - in una beuta da 100 ml porre
- in una beuta da 100 ml porre 


|
Post n°34 pubblicato il 13 Giugno 2010 da paoloalbert
Eccoci alla fase 2, meno impegnativa della prima ma non banale nemmeno questa. 5-nitro-2,3-diidro-1,4-ftalazinadione con la procedura seguente: Materiale occorrente: Occhio all'idrazina ed ai i suoi sali: sono sostanze tossiche e cancerogene, da maneggiare con le dovute cautele!
Scaldare la miscela su bagno di sabbia a 170°-180° (la sabbia ad una decina di gradi in più) per due ore mescolando ogni tanto e poi lasciar raffreddare. Alla fine la miscela è sempre color beige, non è per niente scura come si legge in certe sintesi. Aggiungere 40 ml di acqua, mescolare e filtrare aspirando bene per rimuovere il più possibile la glicerina ed il solfato di sodio formatosi. Lavare il residuo con altre due porzioni da 5 ml di acqua e lasciar asciugare all'aria o meglio in forno a 80-90°.
La resa è di circa 3 g (80%) di 5-nitro-2,3-diidro-1,4-ftalazinadione, che si presenta come una polvere amorfa di colore beige chiaro. |
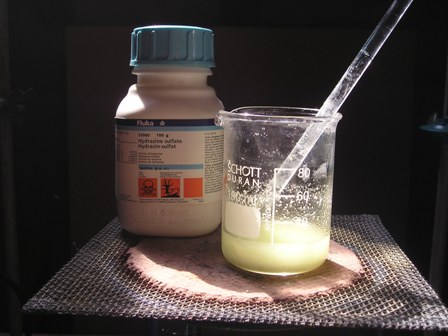 - in un becker da 100 ml porre
- in un becker da 100 ml porre  Aggiungere a questo punto
Aggiungere a questo punto  Quando il residuo è ben secco ma non sovrariscaldato, staccarlo dal vetro del becker al quale può aderire fortemente (io ho usato una spatolina inox ben affilata). Polverizzare il prodotto in un mortaio (sempre guanti!) e porlo in un becker da 50 ml con
Quando il residuo è ben secco ma non sovrariscaldato, staccarlo dal vetro del becker al quale può aderire fortemente (io ho usato una spatolina inox ben affilata). Polverizzare il prodotto in un mortaio (sempre guanti!) e porlo in un becker da 50 ml con 
Inviato da: paoloalbert
il 02/05/2021 alle 21:53
Inviato da: Bepi1249
il 15/04/2021 alle 14:11
Inviato da: Tanner85
il 06/02/2021 alle 00:01
Inviato da: poetryclub
il 04/10/2020 alle 23:00
Inviato da: poetryclub
il 04/10/2020 alle 20:58