CHIMICA sperimentale
Esperienze in home-lab: considerazioni di chimica sperimentale e altro
|
Post n°73 pubblicato il 15 Gennaio 2011 da paoloalbert
Bando alle ciance, oggi lo chef ha un raptus hard e propone a tutti i commensali un piatto di chimicaccia da stomaco buono:
Veniamo subito al sodo, ecco cosa occorre: - in un pallone a due colli da 250 ml con applicato imbuto separatore e termometro che tocca quasi il fondo, introdurre 50 g di naftalina finemente macinata e riscaldare prima fino a fusione (80°) e poi piano piano fino a raggiungere la T di 160° (tolleranza di più o meno 5 gradi). Lasciar raffreddare e versare il prodotto in un becker da un litro contenente 500 ml di acqua, mescolando.
That's all folks! Non è il caso di dire buon appetito... a meno che non ci sia in giro qualcuno che si chiama Eta Beta. |
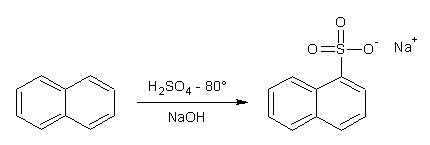
 Lasciando raffreddare si ha abbondantissima separazione di
Lasciando raffreddare si ha abbondantissima separazione di  Il 2-naftalensulfonato sodico
Il 2-naftalensulfonato sodico 
|
Post n°72 pubblicato il 08 Gennaio 2011 da paoloalbert
Se dico "vittoriano", non vien subito da pensare a quel romantico periodo a cavallo tra la prima metà e la fine dell'ottocento? C'è da dire che le pareti delle case borghesi (specialmente inglesi) erano allora spessissimo ricoperte dalla tappezzeria, la cosiddetta "carta da parato": bene, facciamo finta ora che alla padrona di casa di una ipotetica dimora signorile del tempo piacesse una tappezzeria vivace, magari ad artistici fiorami, foglie e fregi come era di moda... Poteva capitare, ed è effettivamente capitato in tante occasioni, che i signori abitanti di quella certa casa cominciassero ad avere strani sintomi, magari all'inizio semplici starnuti, poi tosse, lacrimazione, mal di gola, nausea, coliche, spasmi muscolari, diarrea, depressione, estrema debolezza... La fabbrica della carta da parati aveva usato per i propri pigmenti il bellissimo verde di Parigi (detto anche verde di Schweinfurt) oppure il verde di Scheele, due composti arsenicali allora molto in uso. Qualche formuletta si impone per capire meglio quella vittoriana tappezzeria: - As, sua maestà l'Arsenico, un grigio semimetallo; è sempre lui il colpevole per definizione, è come il maggiordomo dei gialli delle barzellette! Dopo queste amene considerazioni di tossicologia pre-ecologica, quando la sensibilità ambientale era zero con tutte le sue ovvie conseguenze, ce ne sarebbero da fare altre, ma girate di 180 gradi, verso un altro estremismo.
|
|
Post n°71 pubblicato il 04 Gennaio 2011 da paoloalbert
Accendo ora il computer e mi accorgo che il "contachilometri" ha intanto doppiato quella che per me era una lontanissima boa. Non so chi sarà stato il decimillesimo ospite di questo blog nato quasi per caso giusto un anno fa... è certo che per chi trova un po' di diletto anche nello scrivere (oltre che nello sporcare provette!) è una buona soddisfazione constatare che qualcuno apprezza, o almeno partecipa benevolmente a quanto si fa e a quanto si dice. Auguri rinnovati a questo ignoto decimillesimo amico! |
|
Post n°70 pubblicato il 01 Gennaio 2011 da paoloalbert
|

|
Post n°69 pubblicato il 30 Dicembre 2010 da paoloalbert
La chemioluminescenza è una caratteristica di pochissime sostanze che in determinate condizioni emettono radiazione nel visibile per un certo tempo.
Al buio totale, dopo aver abituato gli occhi, mescolare 10 ml della soluzione A e 10 ml della soluzione B e porre la miscela in un cilindro; aggiungere in un colpo mescolando i 10 ml della la soluzione C: risulterà una debole ma visibilissima luminescenza giallastra, che perdurerà per qualche minuto!
Ecco finalmente il cigno promesso... un cigno da chimici naturalmente! Enjoy with lophine! |
 A- Sciogliere 1 g di lophine in 20 ml di etanolo tiepido
A- Sciogliere 1 g di lophine in 20 ml di etanolo tiepido

|
Post n°68 pubblicato il 30 Dicembre 2010 da paoloalbert
Come anticipato a Natale e con mia personale soddisfazione devo tirare in ballo ancora una volta il simpatico Aleksandr P. Borodin, il quale cita nella sua prima relazione scientifica (1859) le ricerche effettuate sull'idrobenzamide e sui suoi derivati amarina e 2,4,5-trifenilimidazolo. Procedura La fase seguente va eseguita in modo opportuno poichè vengono emessi vapori tossici ed irritanti.
La prossima volta vedremo come visualizzare la chemioluminescenza della lophine... ancora un po' di pazienza! |

|
Post n°67 pubblicato il 28 Dicembre 2010 da paoloalbert
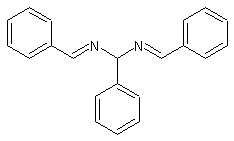
Questo composto (scoperto nel 1836 da A.Laurent, che gli ha dato il nome), è apparentemente una anonima sostanza come migliaia di altre più o meno simili; vedremo invece che l'idrobenzamide possiede una potente chance in più: servì (e servirà a sua volta nella seconda parte di questo lavoro) alla preparazione di un'altra sostanza, decisamente interessante... Ho trovato sul Cumming del 1937 (le fondamenta ciclopiche della chimica sperimentale sono ancorate ai "vecchi" e sacri testi... chi non ricorda il Gattermann di Primo Levi?) una bella sintesina facile facile che ho sperimentato con successo: è una reazione di condensazione e si basa sulla reazione tra l'ammoniaca e la benzaldeide per generare una sostanza che si chiama 1-phenyl-N,N'-bis(phenylmethylidene)methanediamine, e che tutti chiamano molto più amichevolmente hydrobenzamide, oppure, se proprio siamo allergici alla lingua d'oltre Manica, idrobenzamide. Il materiale occorrente è semplice e la sintesi anche, ma già dalla bella formula del prodotto si potrebbe immaginare che esso racchiuda una sorpresina finale; diamo il via dunque alla metamorfosi del brutto anatroccolo, il quale da subito lascia intravvedere con un po' di immaginazione un futuro cigno...
- benzaldeide C6H5-CHO - In una beuta da 100 ml porre semplicemente 10 ml di benzaldeide e 50 ml di ammoniaca concentrata; si forma immediatamente una emulsione bianca; mescolare agitando vigorosamente e ripetere l'operazione ogni tanto per le successive due tre d'ore. L'idrobenzamide si presenta come una polvere bianca (velenosa, classe di rischio T) con odore di benzaldeide (probabile residuo), insolubile in acqua, p.f. teorico 110°, nel mio caso più basso.
Per adesso mettiamo da parte l'anatroccolo; la prossima volta tenteremo di trasformarlo in cigno! Non si vede dalla formula che ha già due bellissime ali? |

|
Post n°66 pubblicato il 22 Dicembre 2010 da paoloalbert
Piccola pausa natalizia dedicata a tutti i visitatori, prendendo spunto dal mio stimato chimico compositore Александр Порфирьевич Бородин (Alieksandr Parfirièvic Baradìn), che presto avrò l'onore di citare ancora una volta riguardo una sintesi che lo toccherà da vicino.
...Buon Natale! |
|
Post n°65 pubblicato il 19 Dicembre 2010 da paoloalbert
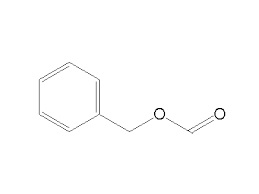
Prima della pausa natalizia voglio completare la trilogia degli esteri benzilici degli acidi grassi inferiori, proponendo la terza e ultima sintesi di un altro estere odoroso importante, quella del formiato.
Materiale occorrente: - In un pallone da 250 ml introdurre 28 ml di alcool benzilico e 46 ml di acido formico all'85%; mescolando aggiungere 2 ml di H2SO4 conc. e predisporre il sistema per riscaldamento a ricadere con mantello riscaldante o con bagno ad olio o sabbia. Ho usato questo sistema, immergendo nella sabbia il termometro. Distillare il prodotto (circa 30 ml) raccogliendo tra 200 e 208°,ottenendo 17 ml di benzile formiato (64%). D. 1,05 - P.e. 203°- Liquido limpido incoloro, oleoso (un po' contro la logica?), con odore fruttato, diciamo una via di mezzo tra il gelsomino e le mandorle, ma più debole e aspro, meno gradevole e diverso dal benzilacetato o propionato.
Sarebbe interessante conoscerne l'effettiva purezza, per verificare la presenza di eventuali prodotti di ossidazione e/o reazioni secondarie... ma ci vorrebbe "un apparecchio con la spina!".
|

|
Post n°64 pubblicato il 12 Dicembre 2010 da paoloalbert
Devo decidermi a mettere ogni tanto qualche intermezzo... in mezzo a queste badilate di chimica, altrimenti si rischia l'indigestione! Spesso quando scrivo qualcosa per questo blog mi ascolto in relax qualche musica preferita; per esempio oggi mi sono ascoltato fra l'altro quel capolavoro di Bach che si trova qui sotto, e che in vicinanza del Natale ci sta a pennello. |
|
Post n°63 pubblicato il 09 Dicembre 2010 da paoloalbert
Oggi faremo un uso della chimica sperimentale molto utilitaristico e pratico; saranno più contenti coloro che di solito non capiscono in queste mie strane riflessioni che gusto ci sia nell'attaccare pezzi di molecola ad un'altra per crearne una terza senza alcun fine pratico, ma solo per farlo così, per pura soddisfazione intellettuale. Oggi, come dicevo, niente di tutto questo! Poichè il mio lab confina in qualche modo anche con la lavanderia di casa, vedendo sullo scaffale una bottiglia di candeggina mi è venuto lo sfizio di verificare in quale percentuale vi fosse contenuto il principio attivo, ovvero l'ipoclorito di sodio NaClO. L'analisi della candeggina (almeno "questa" analisi...) si basa su due reazioni redox; in una reazione redox c'è una sostanza che si ossida e una che si riduce (non sto a dire cosa significa altrimenti non ce la caviamo più...). ClO- + 2 H+ + 2 I- --> Cl- + I2 + H2O Traducendo: l'ipoclorito ossida uno ioduro a iodio e lui si riduce a cloruro. Ecco la seconda: 2 S2O3-- + I2 --> 2 I- + S4O6-- Traducendo: il tiosolfato riduce lo iodio a ioduro e lui si ossida a tetrationato. Poste queste premesse, ho preparato: - una soluzione 0,1 M di tiosolfato di sodio (p.m. 248,10), sciogliendo 2,48 g di Na2S2O3.5H2O in 100 ml di acqua (questa soluzione va fatta esattamente) - una soluzione circa al 5% di acido acetico, diluendo 2,5 ml di CH3COOH in 50 ml di acqua - una soluzione circa al 5% di ioduro di potassio KI, sciogliendone 0,5 g in 10 ml di acqua - una soluzione di amido in acqua, disperdendone 0,1 g in 5 ml di acqua Procedura: - misurare esattamente 10 ml di candeggina e portarli a 100 ml; prendere esattamente 10 ml di questa soluzione e diluirli in un becker con circa 50 ml di acqua - acidificare aggiungendo 10 ml della soluzione di acido acetico - aggiungere circa 5 ml della soluzione di KI; la soluzione assumerà istantaneamente una colorazione marrone, indice che lo ioduro (in eccesso) ha consumato tutto l'ipoclorito ed ha sviluppato la corrispondente esatta quantità di iodio - con una buretta calibrata aggiungere goccia a goccia la soluzione di tiosolfato finchè la colorazione avrà assunto una colorazione giallina (stà diminuendo lo iodio riducendosi a ioduro ed il colore si attenua) - aggiungere qualche goccia della soluzione di amido (la soluzione assume colore violaceo per il complesso che l'amido forma con lo iodio ancora libero) - continuare la titolazione con la buretta, sempre mescolando, finchè la colorazione tende a sparire; il punto di viraggio non è semplice da cogliere, qui è indispensabile un po' di esperienza. Quando il colore tende a sparire vuol dire che non c'è più iodio libero e che tutto il tiosolfato aggiunto si è trasformato in tetrationato. Ora un po' di calcoli... Dall'analisi delle ossidoriduzioni risulta che un equivalente di ipoclorito viene "consumato" da due equivalenti di tiosolfato. Abbiamo fatto una soluzione 0,1 M di Na2S2O3, la quale contiene 15,81 g/l di tiosolfato anidro, ovvero 0,0158 g ogni ml; siccome ad ogni grammo di tiosolfato corrispondono 0,15 g di ipoclorito, ad ogni ml consumato nella titolazione corrispondono quindi 0,0158 x 0,15 = 0,00237 g di NaClO Nel mio caso il punto di viraggio (su tre prove eseguite in sequenza) è stato raggiunto mediamente con 22,5 ml di tiosolfato: 22,5 x 0,00237 = 0,053 Eccoci finalmente al traguardo: Salvo errori ed omissioni, naturalmente! |
|
Post n°62 pubblicato il 05 Dicembre 2010 da paoloalbert
L'altra volta ho parlato in modo discorsivo (e molto incompleto!) degli elementi delle terre rare; oggi l'argomento entra nel particolare della nuda chimica sperimentale e ci entra di brutto. Quanto segue è quindi strettamente riservato agli sporcaprovette impenitenti e amanti degli elementi esotici! Taca banda! |
|
Post n°61 pubblicato il 02 Dicembre 2010 da paoloalbert
Nella splendida biblioteca storica della mia città (non dico quale) ho da poco scoperto che esiste una monumentale opera enciclopedica in francese sulla chimica inorganica che farebbe la felicità di qualsiasi chimico sperimentale. Dopo questa inquietante introduzione, qualcuno (il solito coraggioso...) si chiederà cosa diavolo siano queste Terre Rare, che hanno perfino l'onore della lettera maiuscola. -Scandio, Yttrio |
|
Post n°60 pubblicato il 27 Novembre 2010 da paoloalbert
Mi ricordo che da ragazzo guardavo sempre con intimo sollievo (perchè non era capitato a me!) quelle pitturate di mercurocromo fatte senza economia che arrossavano braccia e gambe di qualche sventurato coetaneo feritosi nei vari modi in cui una volta giocando ci si feriva (oggi il mondo è cambiato anche in tal senso...). Materiali occorrenti:
Lasciare in riposo un paio d'ore, filtrare su buchner e lavare bene prima con poco etanolo poi con acqua. Seccare all'aria il prodotto di addizione eosina.etanolo.
- Mettere in un cristallizzatore una ventina di ml di ammoniaca concentrata.
Questo composto si presenta come una polvere di un bel verde oliva scuro a riflessi metallici (la foto non rende il colore), di enorme potere colorante e solubile in acqua; tracce di esso impartiscono all'acqua la fuorescenza verde rossastra che si vede in foto, ed alle mani delle belle macchie rosse indelebili per qualche giorno!
|
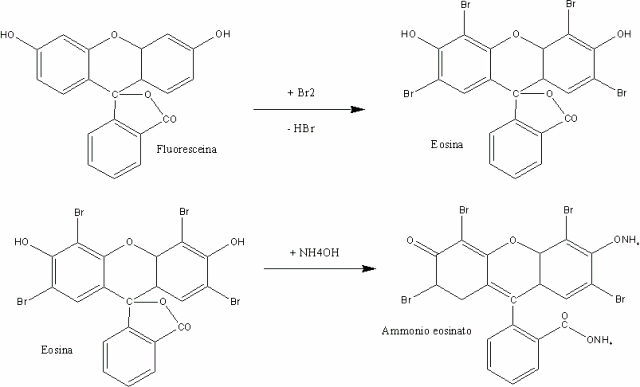
 - Porre in una beuta da 25 ml
- Porre in una beuta da 25 ml  -
-


|
Post n°59 pubblicato il 21 Novembre 2010 da paoloalbert
Tutti gli anni, il lunedì più vicino a ferragosto, da una splendida posizione panoramica collinare a 700 m di altezza e prospiciente la mia visualità estiva, si effettua un tradizionale e seguitissimo spettacolo pirotecnico, che fa alzare gli occhi a migliaia di spettatori raggiunti anche a grande distanza dalle luci e dai colori di questi meravigliosi fuochi.
A parte l'effetto coreografico, come è possibile realizzare questi spettacoli? Come si ottengono queste luci colorate? La base di lancio di ogni fuoco è ancora la gloriosa polvere nera, fatta, come quasi tutti sanno, di una miscela di nitrato di potassio, KNO3, di carbone di legna e di zolfo. Analizziamo ora i colori, che sono quelli che ci interessano di più. Determinante ai fini del colore del fuoco è il comburente o un sale ad esso miscelato: sono in genere sostanze fortemente ossidanti derivate da un metallo i cui sali colorano la fiamma. Luci rosse: sali di stronzio, nitrato Sr(NO3)2, carbonato SrCO3 Luci verdi: sali di bario, nitrato Ba(NO3)2, clorato Ba(ClO3)2 Luci gialle: sali di sodio, ossalato Na2(COO)2, nitrato NaNO3, carbonato Na2CO3 Luci azzurre: sali di rame, carbonato CaCO3, cloruro CuCl2, ossicloruro CuO.CuCl2, solfato ammoniacale CuSO4.nNH4OH. Luci viola: miscele di coloranti rossi e verdi (Sr + Cu + eventuale Hg2Cl2) Luci arancio: miscele di coloranti rossi e gialli (Sr + Na) Luci bianche: alluminio Al, magnesio Mg, siliciuro di calcio CaSi2, antimonio Sb L'ossidante classico era una volta il clorato di potassio KClO3, ora sempre più sostituito dal perclorato KClO4, molto meno pericoloso.
E dopo questo trionfo di sali di stronzio e bario, godiamoci Händel ! |

|
Post n°58 pubblicato il 18 Novembre 2010 da paoloalbert
La sintesi di oggi è una sintesi decisamente estiva, quando lavorare all'aperto, magari nella frescura di un bell'albero ombroso, è una soddisfazione che rende ancora più piacevole il lavoretto che si va a fare... tanto più perchè oggi SI DEVE lavorare all'aperto! Materiali occorrenti: - Potassio iodato KIO3
Sono partito dai 14 g di KIO3 ottenuti per elettrolisi e purificazione come descritto in un post precedente. -Sciogliere lo iodato in 150 ml di acqua tiepida, aggiungere 15 g di KOH e porre la soluzione in un recipiente alto e stretto (ideale un cilindro graduato); pesare ora tutto il sistema.
In un pallone a doppio collo da 250 ml mettere 15 g di TCCA acido tricloroisocianurico ed in un imbuto gocciolatore porre 80 ml di HCl al 15%; far gocciolare molto lentamente l'acido nel TCCA in modo da avere un flusso regolare di cloro, che viene fatto gorgogliare (tre/quattro bolle al secondo) con un opportuno tubo in vetro sagomato sul fondo del recipiente con la soluzione di iodato. 3 KIO3 + 6KOH + 2 Cl2 --> K4I2O9 + 4 KCl + 3 H2O
Il K4I2O9 rimane nella soluzione fortemente alcalina. K4I2O9 + 2 HCl --> 2 KIO4 + 2 KCl + H2O Lavare opportunamente con acqua fredda e lasciar asciugare. Il periodato si presenta sotto forma di bei cristalli bianchi/trasparenti molto pesanti (d.3,62); la solubilità è 8 g/l a 20°-
Ecco il risultato ottenuto.
|
 LA SUCCESSIVA OPERAZIONE VA TASSATIVAMENTE ESEGUITA ALL'APERTO, come si diceva all'inizio.
LA SUCCESSIVA OPERAZIONE VA TASSATIVAMENTE ESEGUITA ALL'APERTO, come si diceva all'inizio.

|
Post n°57 pubblicato il 13 Novembre 2010 da paoloalbert
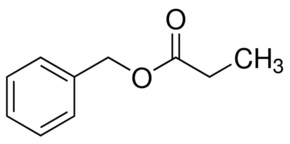
Ogni tanto, irregolare ma prevedibile... zack! : ecco che arriva l'estere! Per la parte generale ved. eventualmente l'acetato.
Materiale occorrente:
- In un pallone da 250 ml introdurre 30 ml di alcool benzilico e 50 ml di acido propionico; mescolando energicamente aggiungere 1 ml di H2SO4 conc. (ho messo volutamente poco acido e preferito giocare sul tempo piuttosto che rischiare ossidazioni inopportune vista la T° elevata in gioco) predisporre il sistema per riscaldamento a ricadere con mantello riscaldante o con bagno ad olio o sabbia. Alla fine separare dall'ultima acqua con imbuto separatore e seccare con 5 g di CaCl2.
Anche il propionato di benzile è un liquido limpido incoloro, leggermente oleoso e con forte odore gradevole aromatico fruttato, molto simile all'acetato ma meno "duro" di questo; è un costituente anch'esso dell'essenza di gelsomino e simili ed è ampiamente usato in profumeria per fragranze di tipo fruttato o orientali. |


|
Post n°56 pubblicato il 11 Novembre 2010 da paoloalbert
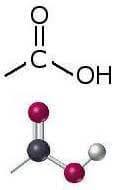
Parafrasando al plurale la celebre frase di Don Abbondio, cosa sono questi acidi carbossilici? Passante, ti è chiara innanzitutto la differenza tra Chimica Organica e Chimica Inorganica? Intanto c'è da dire che gli acidi carbossilici fanno parte della chimica organica e quindi per forza devono contenere il carbonio. L'atomo di carbonio forma dei legami (e qui ci vuole un provvidenziale omissis...) con gli altri atomi adiacenti, formando delle "combinazioni" fisse che originano poi le proprietà della molecola completa.
Quando questa struttura appare in una molecola la fa definire "acido carbossilico", perchè il gruppo -COOH è proprio detto "carbossile". Facciamo qualche esempio pratico: L'acido numero due? Questo tutti lo conoscono perchè è l'acido acetico, costituente fondamentale (diluito al 6%) del normale aceto da tavola, dal quale ne trae il nome. In questo caso la famosa lineetta è collegata con un gruppo -CH3 (detto "metile"), e la formula di conseguenza è CH3-COOH Il terzo acido aggiunge al CH3- anche un -CH2- e quindi la somma totale diventa CH3-CH2-COOH e l'acido originatosi prende il nome di acido propionico (ripeto che c'è un motivo per i nomi, ma non lo dico ora). L'appetito vien mangiando: agggiungiamo ogni volta un -CH2- in più e andiamo avanti con la catenella... CH3-CH2-CH2-COOH: ecco l'acido butirrico... CH3-(CH2)3-COOH: ecco l'acido valerico (o valerianico), e così via. |
|
Post n°55 pubblicato il 06 Novembre 2010 da paoloalbert
Niente paura... non si tratta dei miei compagni di scuola! Ma andiamo con ordine... - Friedrich Wohler: possiamo chiamarlo il padre della chimica organica? E' colui che con uno storico esperimento demolì la secolare Teoria della Forza Vitale, secondo la quale tutte le sostanze organiche hanno origine biologica e che dall'"inorganico" non si può passare all'"organico"! - Peter Griess: per le sue ricerche sull'azoto organico e le diazotazioni è addirittura passato alla storia per il suo celeberrimo reattivo (reattivo di Griess) per la ricerca dei nitriti. - Thedor Curtius: studi fondamentali sulle azidi e l'acido azotidrico; Reazione di Curtius, in cui una acilazide riarrangia in isocianato. - Vladimir Markovnikov: proprio quello della regola che porta il suo nome, e che afferma che nelle reazioni di addizione elettrofila l'idrogeno si lega al carbonio più idrogenato del doppio legame. - Ernst Otto Beckmann: l'inventore del sensibilissimo termometro differenziale per lo studio delle proprietà colligative e della scoperta della Trasposizione di Beckmann, nella quale da una ossima si può ottenere un'ammide. - Carl Graebe: importanti studi sui coloranti, la fucsina, l'alizarina, la purpurina, ecc. - Constantin Fahlberg: scopritore del 1,1-Dioxo-1,2-benzothiazol-3-one, ovvero la saccarina. - Nikolai Menshutkin: al quale si deve l'omonima reazione, nella quale una ammina terziaria viene trasformata in sale di ammonio quaternario per mezzo di un alogenuro alchilico. - Jacob Vohlard: ancora scoperte e importanti ricerche di chimica organica su alogenazioni, aminoacidi, la sintesi del tiofene, la ciclizzazione, determinazioni in chimica analitica. ...e ce ne sarebbero ancora, ma basta, mi fermo qui! Se tali e tanti erano i semplici "compagni di scuola" di Kolbe, per estensione possiamo facilmente immaginare quale fosse l'impegno tedesco nella ricerca universitaria, specificamente chimica, nella Germania di fine ottocento! Quasi quasi verrebbe voglia di paragonarla all'attuale situazione della ricerca italiana... No, fermi con le scarpe! Giù le uova! Ho scherzato! Non tiratemi niente addosso...!!! |
|
Post n°54 pubblicato il 01 Novembre 2010 da paoloalbert
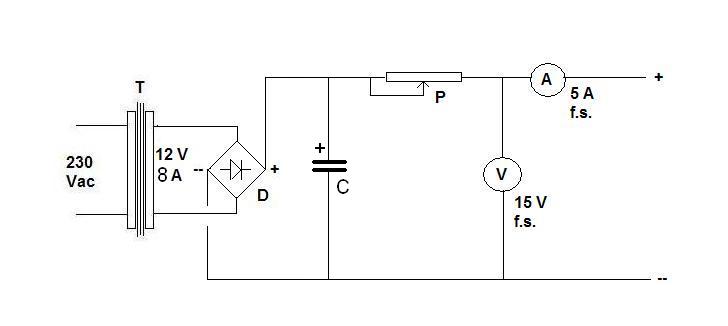
Il 10 aprile 2010 (post n.25) ho presentato la sintesi elettrolitica del bromato di potassio KBrO3; poichè è stato un lavoretto di soddisfazione, ho ripetuto l'esperienza sostituendo lo iodio al bromo e facendo stavolta la sintesi dello iodato di potassio, KIO3.
La fotografia mostra l'alimentazione della celletta con l'accrocco elettrico assemblato in maniera volante e provvisoria, prima di essere debitamente inscatolato per gli usi futuri; in primo piano l'indispensabile amperometro e a destra il potenziometro ceramico a filo per la regolazione della corrente.
L'"incasinamento" del banco di lavoro è dovuto proprio alla sperimentazione pratica dell'alimentatore... la prossima volta sarà molto più ordinato! Materiale occorrente: Potassio ioduro KI Sono partito da 20 g di KI, sciolti in 40 ml di acqua, aggiungendo i soliti 50 mg di bicromato come catalizzatore.
Questi 14 g di KIO3 sono stati destinati a miglior causa, ovvero alla sintesi del periodato di potassio KIO4 per ulteriore ossidazione in altro modo, come si vedrà in un successivo post dedicato. |


Inviato da: paoloalbert
il 02/05/2021 alle 21:53
Inviato da: Bepi1249
il 15/04/2021 alle 14:11
Inviato da: Tanner85
il 06/02/2021 alle 00:01
Inviato da: poetryclub
il 04/10/2020 alle 23:00
Inviato da: poetryclub
il 04/10/2020 alle 20:58