
–
L’EMA e i responsabili delle Agenzie dei medicinali ( HMA ) hanno pubblicato un piano di lavoro sull’intelligenza artificiale ( AI ) fino al 2028, definendo una strategia collaborativa e coordinata per massimizzare i benefici dell’Intelligenza Artificiale ( AI ) per le parti interessate gestendo al contempo i rischi.
Il piano di lavoro aiuterà la rete europea di regolamentazione dei medicinali ( EMRN ) a cogliere le opportunità dell’intelligenza artificiale per la produttività personale, automatizzando processi e sistemi, aumentando la comprensione dei dati e supportando un processo decisionale più solido a beneficio della salute pubblica e degli animali.
Il piano di lavoro sull’intelligenza artificiale, preparato nell’ambito del gruppo direttivo congiunto HMA-EMA Big Data ( BDSG ), garantisce che l’EMRN rimanga in prima linea nel trarre vantaggio dall’intelligenza artificiale nella regolamentazione dei medicinali. Il piano di lavoro è stato adottato dal Consiglio di amministrazione dell’EMA nella riunione di dicembre 2023.
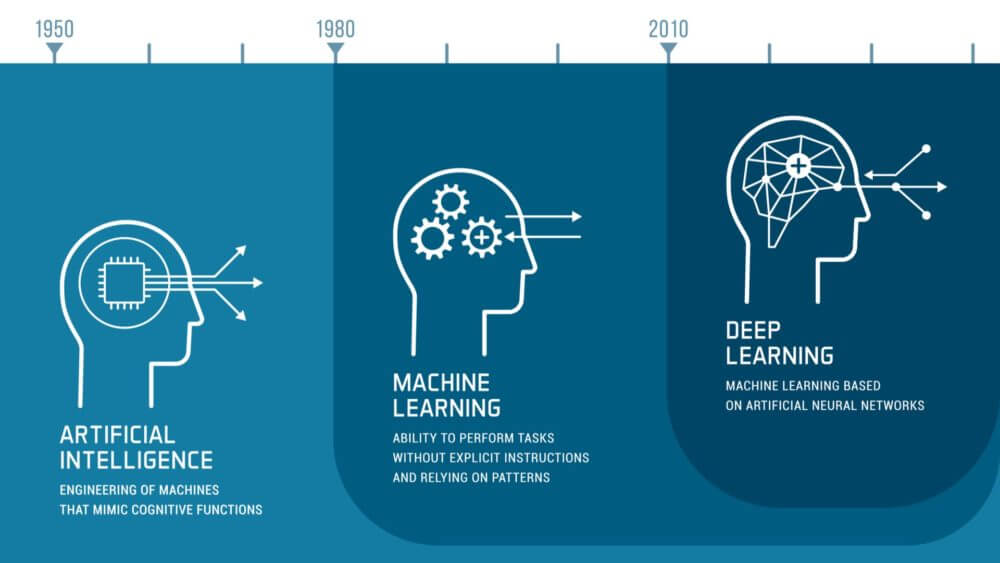
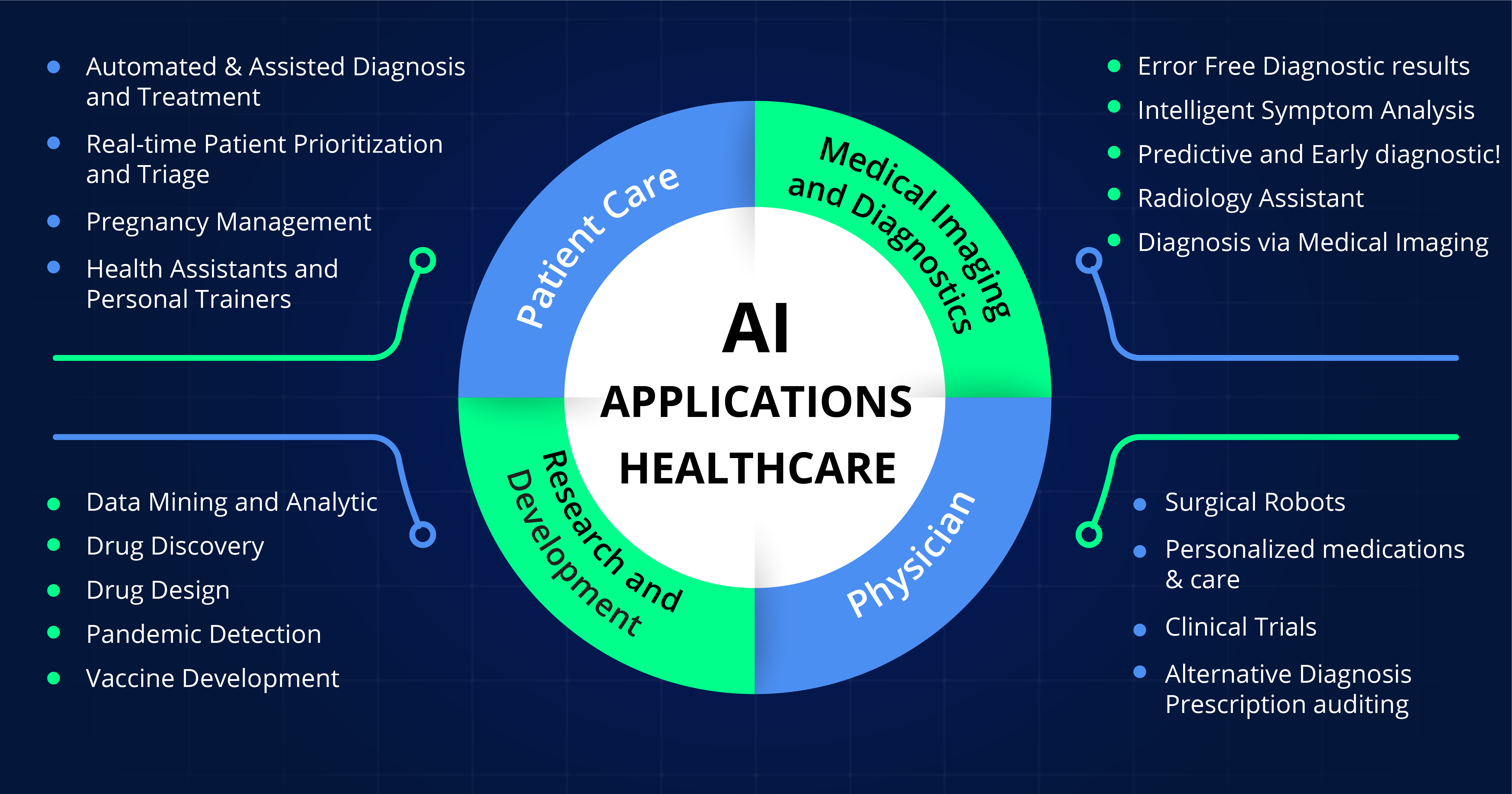
Il campo dell’intelligenza artificiale si sta sviluppando rapidamente. Le aziende farmaceutiche utilizzano sempre più strumenti basati sull’intelligenza artificiale nella ricerca, nello sviluppo e nel monitoraggio dei medicinali. Le autorità nazionali competenti stanno rispondendo alle nuove opportunità e sfide iniziando a utilizzare e sviluppare strumenti di intelligenza artificiale. Il piano di lavoro si concentra su quattro dimensioni chiave:
1) Orientamento, politica e sostegno al prodotto: le azioni si concentrano sul supporto continuo ai prodotti in fase di sviluppo, nonché sullo sviluppo e sulla valutazione di orientamenti adeguati per l’uso di AI nel ciclo di vita di un medicinale. I lavori sono già iniziati con la consultazione pubblica in corso sul documento di riflessione su AI, aperta fino alla fine di dicembre 2023. Inoltre, nel 2024 inizieranno i preparativi per sostenere l’attuazione della legge dell’Unione Europea su AI.
2) Strumenti e tecnologie di intelligenza artificiale: l’obiettivo è identificare e fornire strutture in tutta la rete per utilizzare gli strumenti di intelligenza artificiale per aumentare l’efficienza, migliorare la comprensione e l’analisi dei dati e supportare il processo decisionale. Sarà garantito il pieno rispetto della normativa sulla protezione dei dati.
3) Collaborazione e formazione: le iniziative sono progettate per sviluppare continuamente la capacità della rete, dei partner e delle parti interessate di stare al passo con il campo in evoluzione dell’intelligenza artificiale.
4) Sperimentazione: il piano di lavoro riconosce il ruolo fondamentale della sperimentazione nell’accelerare l’apprendimento e acquisire nuove conoscenze. Vengono proposte diverse azioni per garantire un approccio strutturato alla sperimentazione attraverso la rete.
Poiché la tecnologia dell’intelligenza artificiale è in rapida evoluzione, compresi gli aspetti etici e politici ad essa correlati, il BDSG aggiornerà regolarmente il piano di lavoro. Gli enti regolatori, gli sviluppatori di farmaci, gli accademici, le organizzazioni dei pazienti e le altre parti interessate saranno informati e coinvolti durante tutta l’attuazione del piano.
ENGLISH VERSION
Artificial intelligence workplan to guide use of AI in medicines regulation
EMA and the Heads of Medicines Agencies (HMAs) have published an artificial intelligence (AI) workplan to 2028, setting out a collaborative and coordinated strategy to maximise the benefits of AI to stakeholders while managing the risks.
The workplan will help the European medicines regulatory network ( EMRN ) to embrace the opportunities of AI for personal productivity, automating processes and systems, increasing insights into data and supporting more robust decision-making to benefit public and animal health.
The AI workplan, prepared under the joint HMA-EMA Big Data Steering Group ( BDSG ), ensures the EMRN remains at the forefront in benefiting from AI in medicines regulation. The workplan was adopted by EMA’s Management Board at its December meeting.
The field of AI is developing swiftly. Pharmaceutical companies increasingly use AI-powered tools in research, development and monitoring of medicines. National competent authorities are responding to the new opportunities and challenges by starting to use and develop AI tools. The workplan focuses on four key dimensions:
Guidance, policy and product support: Actions focus on continuous support to products in development as well as the development and evaluation of appropriate guidance for the use of AI in the lifecycle of a medicine. Work has already begun with the ongoing public consultation on the AI reflection paper, open until the end of December 2023. Furthermore, in 2024 preparations to support the implementation of the EU AI Act will start.
A) AI tools and technology: The aim is to identify and provide frameworks across the network to use
B) AI tools to increase efficiency, enhance understanding and analysis of data and support decision-making. Full compliance with data protection legislation will be ensured.
C) Collaboration and training: Initiatives are designed to continuously develop capacity and capability of the network, partners and stakeholders to keep ahead of the evolving field of AI.
D) Experimentation: The workplan acknowledges the fundamental role of experimentation in accelerating learning and gaining new insights. Several actions are proposed to ensure a structured approach to experimentation across the network.
As AI technology is fast evolving, including the ethical and policy aspects related to it, the BDSG will regularly update the workplan. Regulators, medicine developers, academics, patient organisations and other interested parties will be informed and engaged throughout the implementation of the plan.
Source: EMA ( LINK: https://www.ema.europa.eu/en/news/artificial-intelligence-workplan-guide-use-ai-medicines-regulation )
–
MelanomaOnline.net | OncoGinecologia.net | OncoImmunoterapia.net | OncologiaMedica.net | OncologiaOnline.net | TumoriOnline.net | TumoriRari.net






 –
–

