CHIMICA sperimentale
Esperienze in home-lab: considerazioni di chimica sperimentale e altro
|
Post n°429 pubblicato il 05 Gennaio 2020 da paoloalbert
Eh sì, non c'è nulla che non abbia un termine, ed anche questo blog ovviamente ce l'ha. |
|
Post n°428 pubblicato il 21 Dicembre 2019 da paoloalbert
|

|
Post n°427 pubblicato il 12 Dicembre 2019 da paoloalbert
Quand'ero piccolissimo (appena poco più di un bimbo) mio padre mi portò a vedere l'ultima accensione dell'ultima "calcàra" della valle.
La calcàra era una costruzione circolare del tutto simile ad un piccolo nuraghe sardo, alta circa quattro metri, fatta di grosse pietre al contorno e con una grande cavità all'interno alla quale si accedeva tramite una piccola apertura alla base. |

|
Post n°426 pubblicato il 03 Novembre 2019 da paoloalbert
Cosa sarà mai questo strano oggetto qui sotto?
6- Alla fine della piattina di discesa (allora non c'erano i cavi coassiali e dall'antenna agli appartamenti si scendeva con una selva di "piattine", linee bifilari in polietilene) il radiotecnico monta dietro il televisore uno scatolino detto "demiscelatore" che manda i segnali televisivi metà alla vecchia presa d'antenna e metà all'oggetto misterioso protagonista di questa storiella. 8- Acceso il Grundig monumentale, si sposta l'interruttore sporgente dal buco laterale da "1" a "2" e si gira lentamente (lentamente, ragioniere!) la manopola fino a sintonizzare il segnale, che il radiotecnico assicura dover arrivare abbastanza bene nella zona. Era un oggetto all'avanguardia per quei tempi perchè le frequenze in gioco molto alte obbligavano all'uso di transistor speciali (nel convertitore di cui sopra i famosi AF239 e AF139).
|


|
Post n°425 pubblicato il 03 Novembre 2019 da paoloalbert
Allora andiamo avanti con il nostro "Autan"?
Quando tutta l'ammina è stata immessa, mescolare per una decina di minuti.
Quando feci la sintesi avevo scritto: -"ora che abbiamo fatto l'Autan originale, la DEET è stata sostituita commercialmente con l'icaridina. Che ci tocchi fare la sintesi anche di questa?"
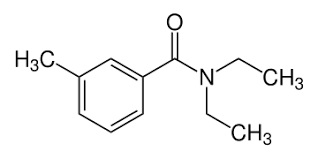
Ma la dietiltoluamide non è incolora? Certo che lo è, ma se l'avessi trattata con carbone attivo sarebbe stata bella ma me ne sarebbe rimasta troppo poca. Mi accontentai. |

 Separare la fase eterea superiore, lavarla con un po' di soluzione di NaOH e riseparare.
Separare la fase eterea superiore, lavarla con un po' di soluzione di NaOH e riseparare. Eliminare l'etere in eccesso mediante evaporazione; io ho lasciato la soluzione in recipiente aperto e arieggiato fino a che tutto l'etere è evaporato.
Eliminare l'etere in eccesso mediante evaporazione; io ho lasciato la soluzione in recipiente aperto e arieggiato fino a che tutto l'etere è evaporato.
|
Post n°423 pubblicato il 23 Settembre 2019 da paoloalbert
Tutti ci siamo spalmati sulla pelle almeno una volta, chi più chi meno, la dietiltoluamide dell'Autan, simbolo commerciale di un famoso repellente per insetti. Ora questa sostanza è stata sostituita dalla icaridina ma la dietiltoluamide rimane in memoria come il repellente-tipo.
DEET Icaridina
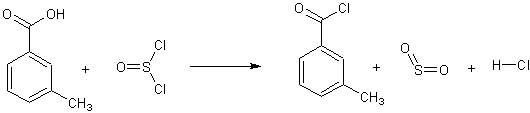
Ho provato (ormai qualche anno fa, ma la pubblico adesso per i motivi che dirò) a farne la sintesi, con maggior interesse del solito perchè la stessa è tosta e quindi di particolare soddisfazione una volta arrivati alla fine. Ecco la reazione:
La procedura DEVE essere eseguita con tutti i sottintesi ai quali ho accennato prima, sotto cappa o in condizioni opportune.
Lasciar raffreddare e tenere il cloruro di m-toluile prodotto (ermeticamente chiuso con tappo smerigliato) per la successiva reazione con la dietilammina.
Ecco il cloruro di toluile! Conclusione la prossima volta. |
 Mettere in un palloncino da 100 ml 10 g di
Mettere in un palloncino da 100 ml 10 g di 

|
Post n°422 pubblicato il 09 Giugno 2019 da paoloalbert
Lo sconsolato post di oggi è purtroppo un altrettanto sconsolato aggiornamento al post n. 350 ...°°°OOO°°°... Tutto quanto sopra lo potrei riportare pari-pari (fatte le debite proporzioni!!!) per il Museo della Scienza e della Tecnica di Milano.
|
|
Post n°421 pubblicato il 19 Aprile 2019 da paoloalbert
Che domanda! Ovvio che lo contengono, lo sanno anche i bimbi.
In casi come questo di piccole quantità di sostanza è molto conveniente separare per centrifugazione il precipitato anzichè perderne gran parte sul filtro. Procedendo con il metodo descritto dal Treadwell, si tratta il precipitato con una soluzione di cloruro di sodio, nella quale si scioglie perfettamente con formazione di HgCl2 e acido acetondicarbossilico, il quale, come gli altri composti chetonici o fenolici, si colora in rossastro in presenza di cloruro ferrico.
L'immagine mostra a destra la soluzione ferrica, al centro la prova in bianco con tre gocce di FeCl3 e a sinistra la colorazione rossa indice della presenza di quell'acido acetondicarbossilico formatosi per ossidazione dal citrico. |


|
Post n°420 pubblicato il 13 Marzo 2019 da paoloalbert
Lungo una strada montana dei miei posti è oggi tutto un fiorire di primule gialle; se fossero usignoli l'ambiente rimbomberebbe di inni alla primavera.
Si tratta della diffusissima Primula vulgaris che vive in tante parti delle nostre colline; durante l'anno si fa notare con difficoltà solo per qualche fogliolina verde rugosa, mentre esplode nel suo breve fulgore all'inizio della primavera.
|

|
Post n°419 pubblicato il 05 Febbraio 2019 da paoloalbert
C'è nella profonda Milano nord, alla Comasina, una anonima (nel senso di poco conosciuta) via intitolata a Gerolamo Forni, farmacista milanese del XIX-esimo secolo. |
|
Post n°418 pubblicato il 16 Gennaio 2019 da paoloalbert
Mi diceva ieri la Ŝiura Gina: -Sai che mi salta il ticchio di fare un esperimento?- |
|
Post n°417 pubblicato il 31 Dicembre 2018 da paoloalbert
Stavolta non voglio dire di qualche esotico metallo che arriva tutto (o quasi) da quell'unica miniera situata, di regola, nel posto più scomodo al mondo che uno possa immaginare. Ma nel caso italiano parliamo di un'industria per lo più locale, di alto livello e che non è destinata ad invadere i supemercati e le tabaccherie del mondo e finire magari in mano a gente di dubbia intelligenza che si spara il petardo in mano, come domani mattina puntualmente verificheremo al Telegiornale. E mi va di citare l'azienda francese Ruggieri, che da secoli organizza fuochi degni di un Re, come quelli commissionati nel 1749 da Giorgio II d'Inghilterra e per quali Friedrich Haendel musicò la meravigliosa suite Royal Fireworks Music. Tutto dovrebbe finire nelle mani giuste: guarda di che meravigliose opere d'arte son capaci i cinesi con i loro fuochi...
e tanto per finire veramente in gloria ecco un frammento dei Reali Fuochi d'Artificio di Haendel:
e tanto basta per augurare a tutti Buon DueMilaDiciannove! |

|
Post n°416 pubblicato il 09 Dicembre 2018 da paoloalbert
C'è un'interessante faccenda di cui voglio dire, che non conoscevo e che è molto articolata; la pongo per punti per semplificare più che posso.
A- Ho detto problemi lontani... infatti la Groenlandia è lontana anni luce dai sentimenti italiani. K- Che siano dollari, euro, yuan poco importa, sono QUESTI E SOLO QUESTI (i danè) che contano, il resto son minchiatine. |
|
Post n°415 pubblicato il 14 Novembre 2018 da paoloalbert
La prima volta che son capitato da quelle parti, la città si chiamava Leningrado ed il Krassin stava ancora cercando gas e petrolio nei mari artici e perciò mi sfuggì.
Dal mio blog ogni tanto salta fuori il mio interesse e passione per le spedizioni polari che fecero storia a cavallo tra l'ottocento e i primi decenni del secolo scorso.
E per concludere... ecco PaoloAlbert tutto preso con il timone del Krassin... non è propriamente la plancia com'era ai tempi del Prof. Samoilovic, ma non si può aver tutto! |


|
Post n°414 pubblicato il 18 Ottobre 2018 da paoloalbert
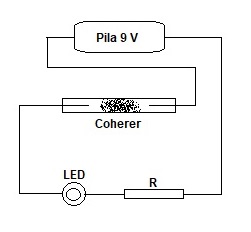
Basta cercare in rete la parola coherer e si trova tutto quello che si vuole su questo misterioso dispositivo elettronico.
Ed ecco la realizzazione pratica, fatta in un quarto d'ora per provarlo. Coherer spento Coherer acceso
In basso si vede la piccola capsula piezo, recuperata da un accendino, che serve a innescare il dispositivo a qualche distanza.
-Il principio di funzionamento del coherer è tuttora non chiaro. |


Inviato da: cassetta2
il 20/04/2025 alle 11:25
Inviato da: paoloalbert
il 02/05/2021 alle 21:53
Inviato da: Bepi1249
il 15/04/2021 alle 14:11
Inviato da: Tanner85
il 06/02/2021 alle 00:01
Inviato da: poetryclub
il 04/10/2020 alle 23:00